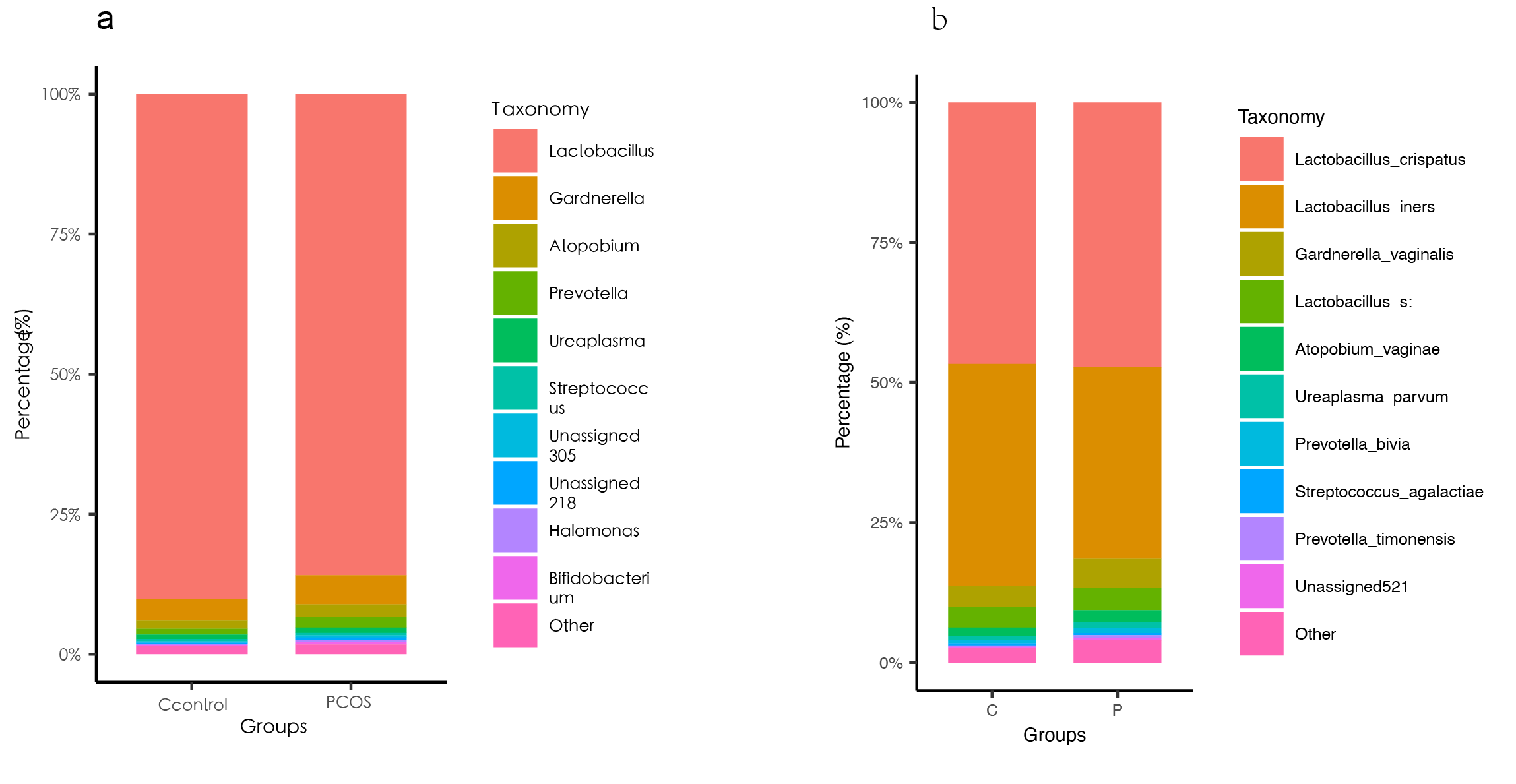
Abstract Objectives To investigate the vaginal microbiome (VMB) of a large sample size of polycystic ovary syndrome (PCOS) patients using 16S ribosomal RNA (16S rRNA) sequencing. Design A cross-sectional study Setting Ji Nan, China Sample A total of 1,446 subjects were recruited (PCOS, n=713, the controls, n=733). Methods Vaginal swabs were analyzed using 16S rRNA gene sequencing. Main outcome measures The microbiome diversity and composition of the PCOS group and control group were compared. In the PCOS prediction model, microbial interaction networks and functions prediction were investigated. Results The PCOS group had a higher alpha diversity in the VMB than controls (P<0.05), while higher intra-group variability was observed in PCOS (P<0.05). At the genus level, the proportion of Lactobacillus in the PCOS group decreased, while the proportion of Gardnerella and Ureaplasma increased (FDR<0.2). Gardnerella vaginalis, Prevotella buccalis, and Prevotella timonensis were identified as differential species and were strongly associated with blood parameters of PCOS. The VMB interaction network indicated that Prevotella and Lactobacillus may be key drivers in the PCOS group. Overall, 55 differentially predicted genes were found between PCOS and controls (FDR<0.25). Conclusions The PCOS group had a higher diversity in the vaginal microbiome and showed an enhanced level of heterogeneity. The proportion of Lactobacillus in PCOS group decreased, while the proportions of Gardnerella and Ureaplasma increased. These results warrant further research that can validate the correlation between PCOS and VMB.