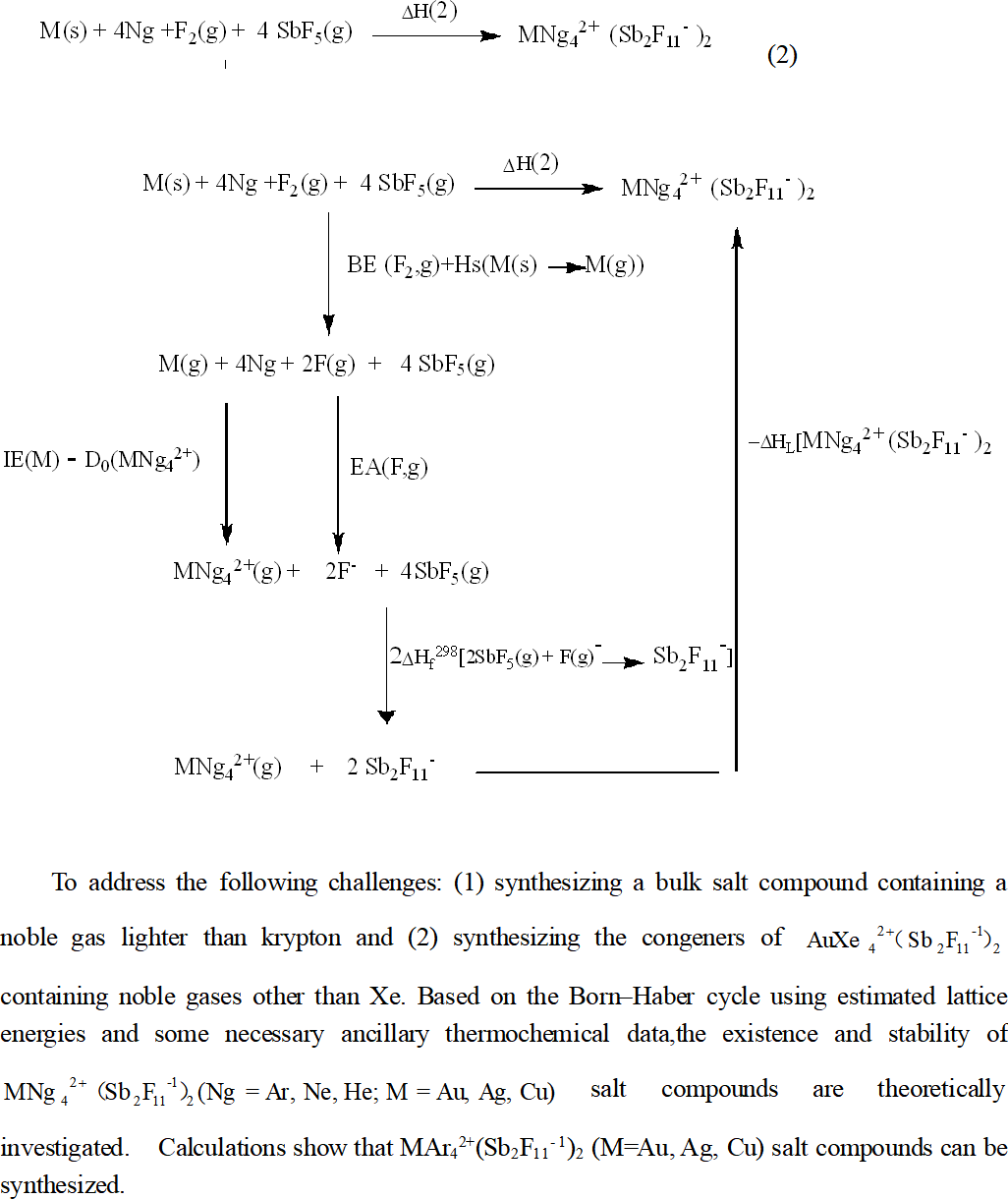
The existence and stability of MNg42+(Sb2F11−1)2 (Ng=Ar,Ne,He,M=Au, Ag, Cu) salt compounds are theoretically investigated in this study. This undertaking is carried out to address the following challenges: (1) synthesizing a bulk salt compound containing a noble gas lighter than krypton and (2) synthesizing the congeners of AuXe42+(Sb2F11−1)2 containing noble gases other than Xe. The reliability of our calculations on the MNg42+(Sb2F11−1)2 (Ng=Ar,Ne,He,M=Au, Ag, Cu) systems is assessed by benchmark calculations of the well-known AuXe42+(Sb2F11−1)2 salt. In the benchmark calculations, a two-pronged evaluation strategy, including direct and indirect evaluation methods, is used to theoretically investigate the spectroscopic constants of AuXe42+and the existence and stability of the AuXe42+(Sb2F11−1)2 salt. The validity of the theoretical calculation methods in the benchmark calculations of AuXe42+(Sb2F11−1)2 allows us to adopt a similar methodology to effectively predict the existence and stability of MNg42+(Sb2F11−1)2 (Ng=Ar,Ne,He,M=Au, Ag, Cu) salt compounds. Calculations based on the Born–Haber cycle using estimated lattice energies and some necessary ancillary thermochemical data show that MAr42+(Sb2F11−1)2 (M=Au, Ag, Cu) salt compounds can be synthesized. The upper-limit stable temperatures are estimated to be −224.43, −146.21, and −80.39 °C. The CuAr42+(Sb2F11−1)2salt compound is a promising candidate. Our calculations also show that the MNg42+(Sb2F11−1)2 (Ng=Ne,He,M=Au, Ag, Cu) salt compounds cannot be stabilized.