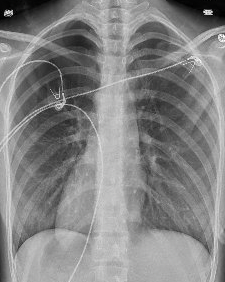
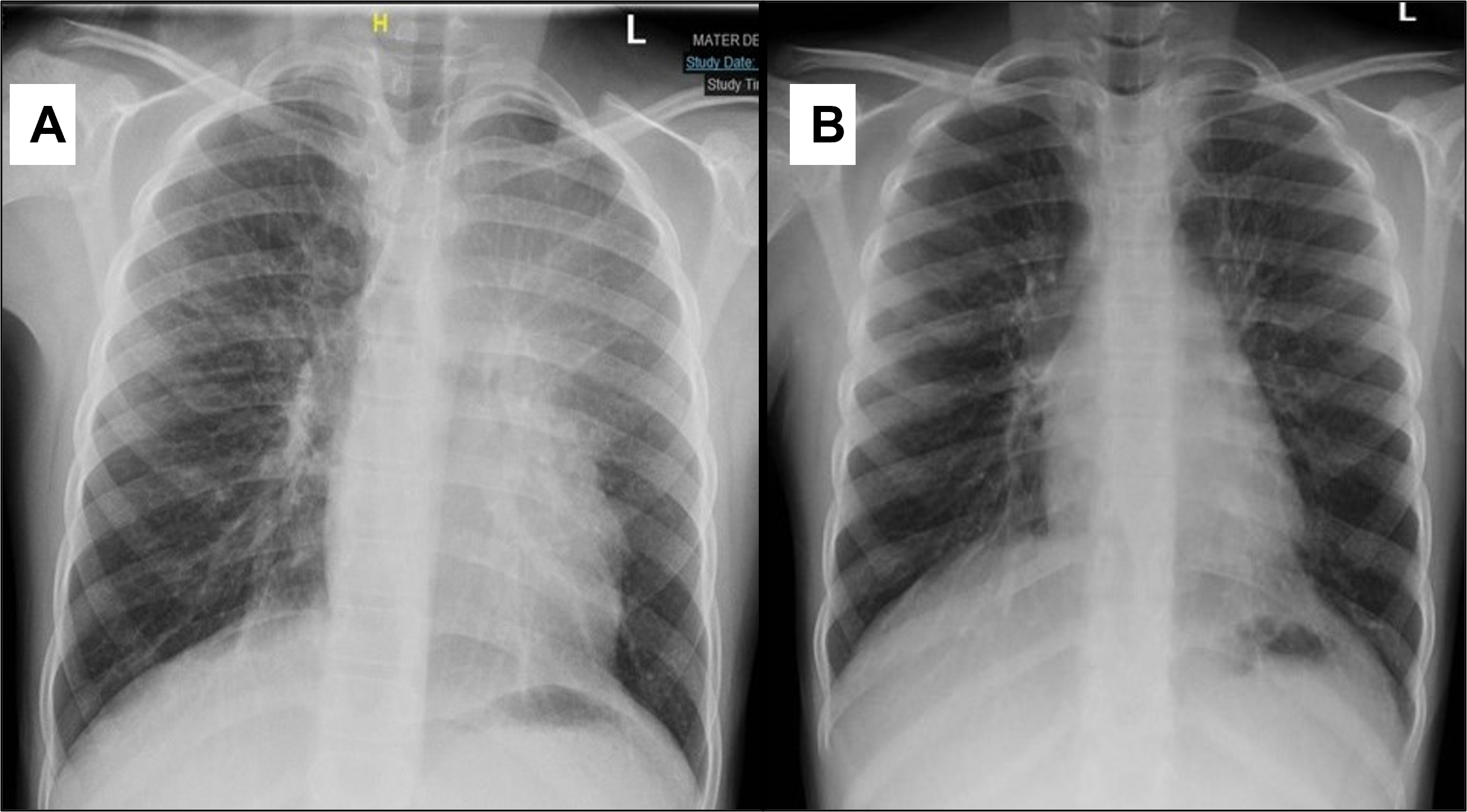
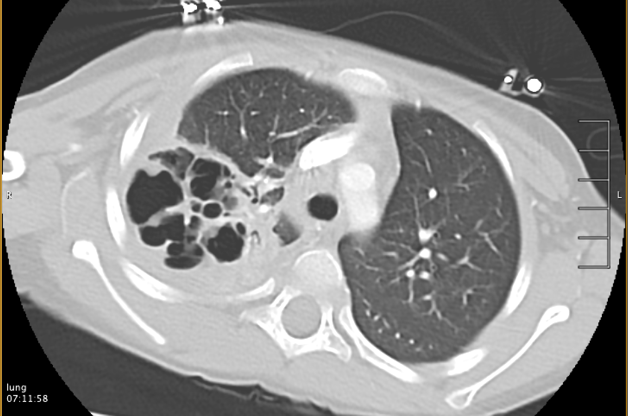
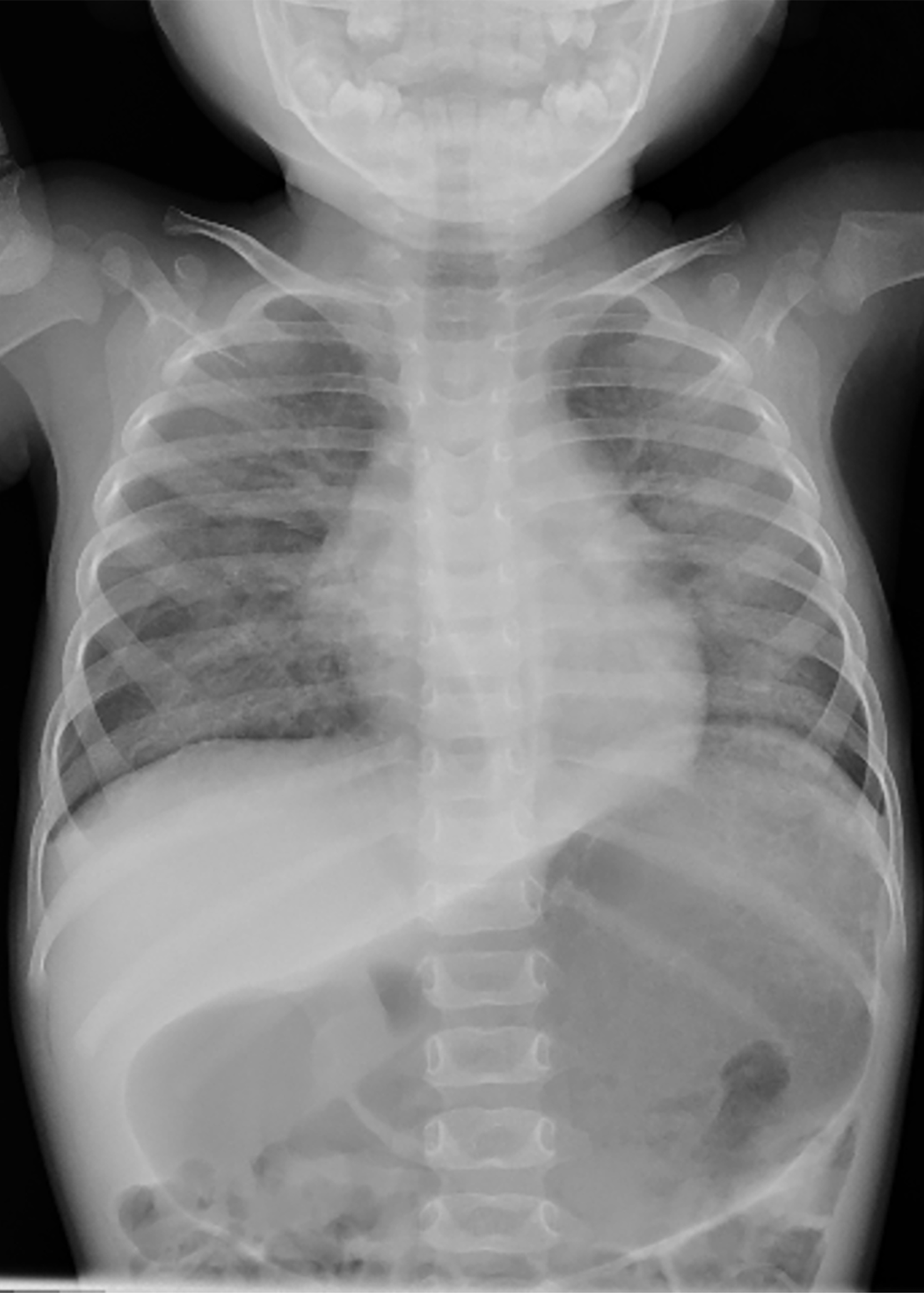
Pediatric Pulmonology , LetterElevated serum TARC/CCL17 levels are associated with childhood interstitial lung disease in patients with SFTPC gene mutationYuto Otsubo MD1; Yuji Fujita MD1; Yusuke Ando MD, PhD1; George Imataka MD, PhD1; Shigemi Yoshihara MD, PhD11Department of Pediatrics, Dokkyo Medical University, 880, Kitakobayashi, Mibu, Shimotsuga, Tochigi, 321-0207, JapanCorresponding author:Yuto OtsuboDepartment of Pediatrics, Dokkyo Medical University, 880, Kitakobayashi, Mibu, Shimotsuga, Tochigi, 321-0293, JapanTel: +81-0282-86-1111Fax: +81-0282-86-7152E-mail: [email protected]:Childhood interstitial lung disease; TARC/CCL17; SFTPCRunning head:Childhood ILD and increased TARC/CCL17 levelTo the Editor,Childhood interstitial lung disease (ILD) is a serious and often life-threatening disease that causes interstitial lung lesion formation during childhood. Several causative genes of childhood ILD, such asSFTPC , SFTPB , and ABCA3 , have been identified in some children. The pathogenesis of ILD caused by SFTPC mutations may include the accumulation of surfactant protein C (SP-C) in vesicles, inhibition of pulmonary surfactant reuptake, and decreased secretion of SP-C, but this remains to be confirmed.1Thymus and activation-regulated chemokine/C-C motif chemokine ligand 17 (TARC/CCL17) is a known disease marker of atopic dermatitis. Recently, an association between idiopathic pulmonary fibrosis, a representative disease with pulmonary interstitial lesions, and TARC/CCL17 has been reported,2 but none between childhood ILD and TARC/CCL17.Here we report our experience of a case of childhood ILD in which the patient had an SFTPC mutation and an elevated TARC/CCL17 level at disease onset that decreased as the patient improved with treatment. TARC/CCL17 may be involved in the pathogenesis of ILD in children withSFTPC mutation, which is different from the pathogenesis of atopic dermatitis.An otherwise healthy 15-month-old girl was admitted to our hospital with fever, difficulty breathing, and poor oral intake. Nine days prior to the visit, nasal discharge and cough appeared and gradually worsened; subsequently, poor oral intake appeared. No fine crackles were noted. The patient required supplemental oxygen and was admitted to the hospital. She had no history of respiratory impairment at birth, but she had a family history of ILD in her maternal grandmother. Informed consent was obtained from the patient’s guardians for the publication of this case report.Laboratory tests showed a white blood cell count of 941 × 109/L, neutrophil count of 58%, C-reactive protein level of 0.01 mg/dL, lactate dehydrogenase level of 929 IU/L, Krebs von den Lungen-6 (KL-6) level of 909 U/mL (normal range < 500 U/mL), surfactant protein A level of 2770 ng/mL (normal <43.8 ng/mL), and surfactant protein D level of 319 ng/mL (normal <43 ng/mL), which were suspicious findings for ILD (Table 1). ß-D-glucan level was 7.9 pg/mL (normal <20 pg/mL) and cytomegalovirus antibodies were negative for both IgG and IgM. Chest radiography showed bilateral frosted shadows (Fig. 1), and a chest computed tomography scan showed bilateral diffuse frosted shadows (Fig. 2), leading to ILD diagnosis.Although prednisolone was started on the 2nd day of admission, the patient’s respiratory status did not sufficiently improve, and oxygen supplementation was required. Therefore, methylprednisolone (30 mg/kg/day) was administered twice for 3 consecutive days on days 13-15 and 19-21; however, the respiratory status remained poor. Hydroxychloroquine (10 mg/kg/day) was then started on day 21, and azithromycin (10 mg/kg/day) three times a week was started on day 56, following which the respiratory status gradually improved. On drinking cold water, the patient often coughed and sometimes vomited. When the water was warmed from 4°C to approximately 20°C, coughing and cough-induced vomiting drastically decreased. On day 66 of hospitalization, the patient was discharged with home-based oxygen. Respiratory status, oxygenation, and laboratory data for ILD markers such as KL-6 gradually improved (Table 1).Genetic analysis of SFTPB , SFTPC , ABCA3 ,CSF2RA , and CSF2RB showed SFTPC mutation and p.I73T (c.218T>C), and the ILD was determined to be caused by theSFTPC mutation.Additional examination revealed elevated TARC/CCL17 level at 10,270 pg/mL (normal <998 pg/mL), but IgE (24.2 IU/mL; normal <173 IU/mL) and IL-4 (2.7 pg/mL; normal <6 pg/mL) levels were not elevated in the early stages of treatment. TARC/CCL17 level decreased to 2,122 pg/mL on day 131 after the admission. Granulocyte-macrophage colony-stimulating factor (GM-CSF) level was measured twice on days 11 and 83, and on both, the GM-CSF level was under 5 pg/mL without significant elevation. Anti-GM-CSF antibody (0.3 U/mL; normal <1.7 U/mL) was negative.It has been hypothesized that many of these effector cell populations are recruited by TARC/CCL17 and act profibrogenically, but the details remain largely unknown.3) SFTPC mutations increase the number of abnormal alveolar type 2 epithelial cells (AT2) due to impaired metabolism of SP-C. A knock-in mouse model capable of regulating the expression of an isoleucine-to-threonine substitution at codon 73 (p.I73T) in SFTPC, at the same site as in the present case, showed persistently elevated TARC/CCL17 level in the bronchoalveolar lavage fluid (BALF). Furthermore, the same study also reported that TARC/CCL17 is specifically released by AT2.3 In ILD caused by SFTPC mutations, its pathogenesis involves AT2 hyperplasia. The decrease in TARC/CCL17 level in our patient suggests that either AT2 itself or TARC/CCL17 production from AT2 itself decreased with treatment. In this case, we report, for the first time, elevated serum TARC/CCL17 level in a patient with SFTPC mutation, which decreased with treatment.Our patient showed no symptoms of atopic dermatitis, and she had no skin condition, and no elevation of IgE and IL-4 levels. In the SFTPC p.I73T mouse model mentioned above, no significant level of IL-4 or IL-13 was detected in BALF, and no involvement of the Th2 response was observed.This high TARC/CCL17 level was not considered to be a result of the GM-CSF cascade. TARC/CCL17 is released from macrophages as a product of the GM-CSF cascade.4) GM-CSF is also known to be produced by AT2.5) However, in this case, serum GM-CSF levels were normal and not elevated, both at the beginning of treatment and after improvement.The limitation of this case is that bronchoalveolar lavage was not performed; therefore, the evaluation was based on serum level rather than local lung findings.This case suggests that TARC/CCL17 is involved in ILD pathogenesis. Further elucidation of the chemokine and receptor signaling cascade may lead to the targeting of some stages for therapy, which may be an important issue for future medical treatment. Therefore, elucidation of this pathogenesis is desirable.References1) Beers MF, Mulugeta S. Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu Rev Physiol 2005;67:663-696.2) Sivakumar P, Ammar R, Thompson JR, Luo Y, Streltsov D, Porteous M, McCoubrey C, Ill EC, Beers MF, Jarai G, et al. Integrated plasma proteomics and lung transcriptomics reveal novel biomarkers in idiopathic pulmonary fibrosis. Respir Res 2021;22;273:1-13.3) Nureki SI, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, Barrett M, Nguyen V, Kopp M, Mulugeta S, et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest 2018;128:4008-4024.4) Hamilton JA. GM-CSF-dependent inflammatory pathways. Front Immunol 2019;10;2055:1-8.5) Woo YD, Jeong D, Chung DH. Development and functions of alveolar macrophages. Mol Cells 2021;44:292-300.Conflicts of interest:The authors disclose no conflicts.Contributors:YO cared for the patient, conceived the concept of the case report, and drafted the initial manuscript. YF, YA, GI, and SY critically reviewed the manuscript for intellectual content. All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work. All authors have read and approved the final manuscript.Acknowledgment:We would like to thank Editage (www.editage.com) for English language editing.We thank Dr. Goro Koinuma, Division of Pulmonology, National Center for Child Health and Development, Tokyo, Japan, for his invaluable expert opinion regarding the diagnosis and treatment of the patients.Sources of funding:No funding was obtained for this study.