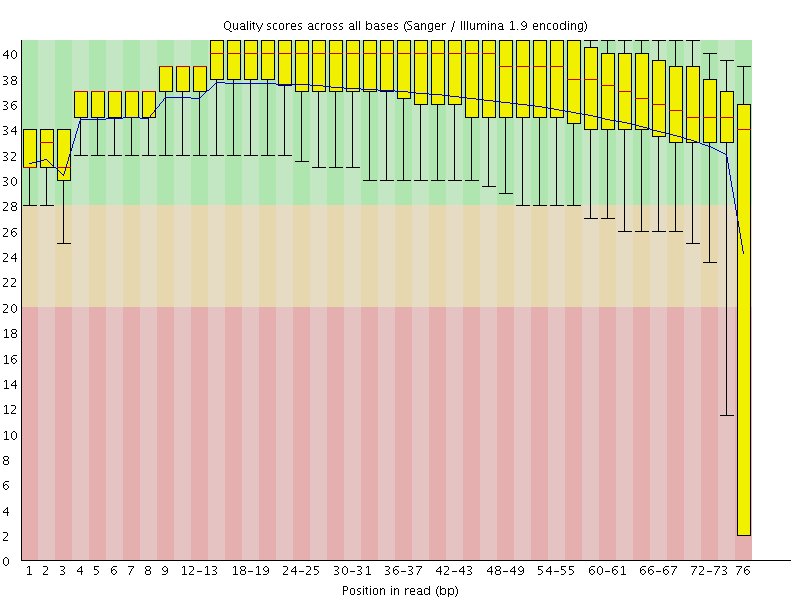
I seek to explore various frequentist and Bayesian methods for drawing conclusions about genetics of gene regulation, both for cis- and trans- association studies, in the context of RNA-seq dataset with known/inferred confounders.IntroductionIn their paper Bayesian new statistics: https://link.springer.com/article/10.3758/s13423-016-1221-4, Kruschke and Liddell explain the divide in the statistical community, between frequentist and Bayesian methods, and between estimation and quantification of uncertainty. Not only do the authors recommend a shift from the former to the latter in both respects, they also try to point out that additional aspects of statistical analyses such as meta-analysis, randomized controlled trials and power analysis may benefit from this shift.Being aware of this recommendation, and also of the recent advances in deep sequencing projects that provide rich information about variation in genotypes, expression and other phenotypes of interest in a variety of settings, I would like not only review, but also further develop, methods that seek to reason about this transition in genomics. As a first step, I focus on RNA-seq studies with genotyping information, which provide a proxy to overall expression levels of proteins and thus cellular activity in a system of interest, and also the potential to separate out genetic and non-genetic basis of variation in expression, giving us important insights into regulatory mechanisms of these systems.Primer: RNA-seq workflow, problems and (partial) solutionsIn this primer, I go through our current (and some not yet implemented) RNA-seq workflow, organizing them into problems and (partial) solutions. In cases where the solution is insufficient (which is true in most cases), I've marked them as possible paper topics.dbGaP - https://www.ncbi.nlm.nih.gov/gapSRA library - https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=run_browserFirst, we can take the sequence library form an experiment, usually in *.fastq format, from public studies shared in the above resources, or from a proprietary dataset. We are currently working with Illumina 76-bp paired-end RNA-seq reads in the case of GTEx data, but certainly are not limited to this setup.Problem:We need to make sure the that the reads are not contaminated (adapter sequences, other non-human reads, etc.), have no unexpected biases, and that the reads have sufficient quality score. In addition, usually the sequences towards the end have poor read quality, and we want to be able to account for this.A solution:Use FastQC and Trimmomatic to perform QC and trim reads for poor quality ends:FastQC manual: https://biof-edu.colorado.edu/videos/dowell-short-read-class/day-4/fastqc-manualTrimmomatic manual: http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/TrimmomaticManual_V0.32.pdfOptional: An example FastQC output from a real GTEx sample: