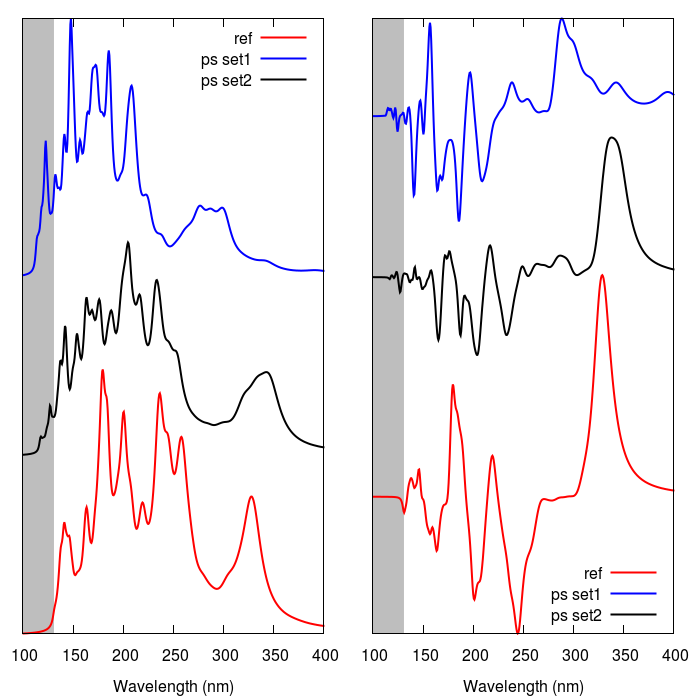
Building on a previous work, pseudopotential sets are developed and tested for a variety of \(sp^2\) and \(sp^3\) carbon fragments. These fragments contain only one or two explicit protons and electrons, and make use of non-atom-centred potentials. They are tested with Density Functional Theory calculations in a selection of chemical environments in which several physical characteristics, including orbital and first ionisation energies, are found to be well-reproduced. They are then employed in the reproduction of molecular absorption spectra for large organic molecules and carbon allotropes, and are found to recreate both absorption and ECD spectra to a high accuracy. They are also found significantly to increase the computational efficiency of TDDFT calculations in which they are used.