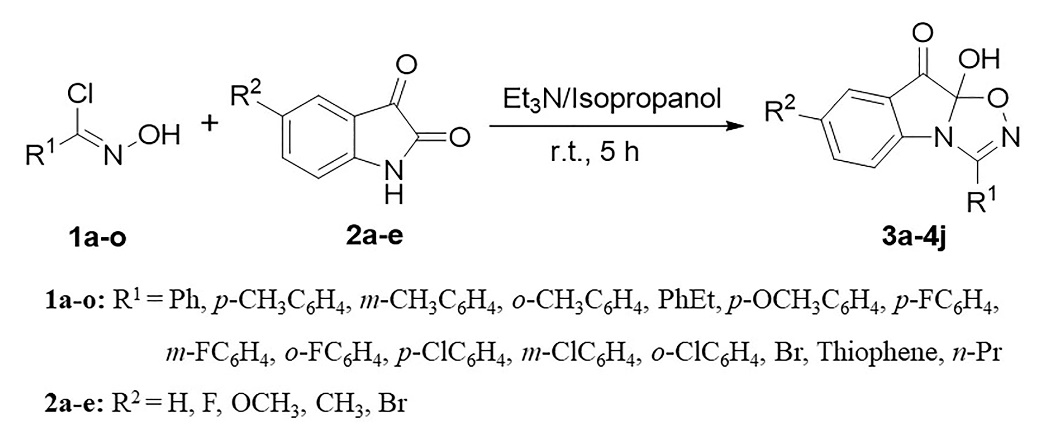
In this study, we performed density functional theory calculations using the B3LYP, M052X, M062X, and APFD functionals to investigate substituent effects on the mechanism of 1,3-dipolar cycloaddition, a classical and effective method for the synthesis of heterocyclic compounds. The results showed that changing the substituents on the chloroxime compounds affects the energy level of the highest occupied molecular orbital and consequently, the progress of the reaction. Finally, it provided an effective idea for this kind of reaction in the design of organic synthesis and the necessary theoretical basis for revealing the course of this reaction.