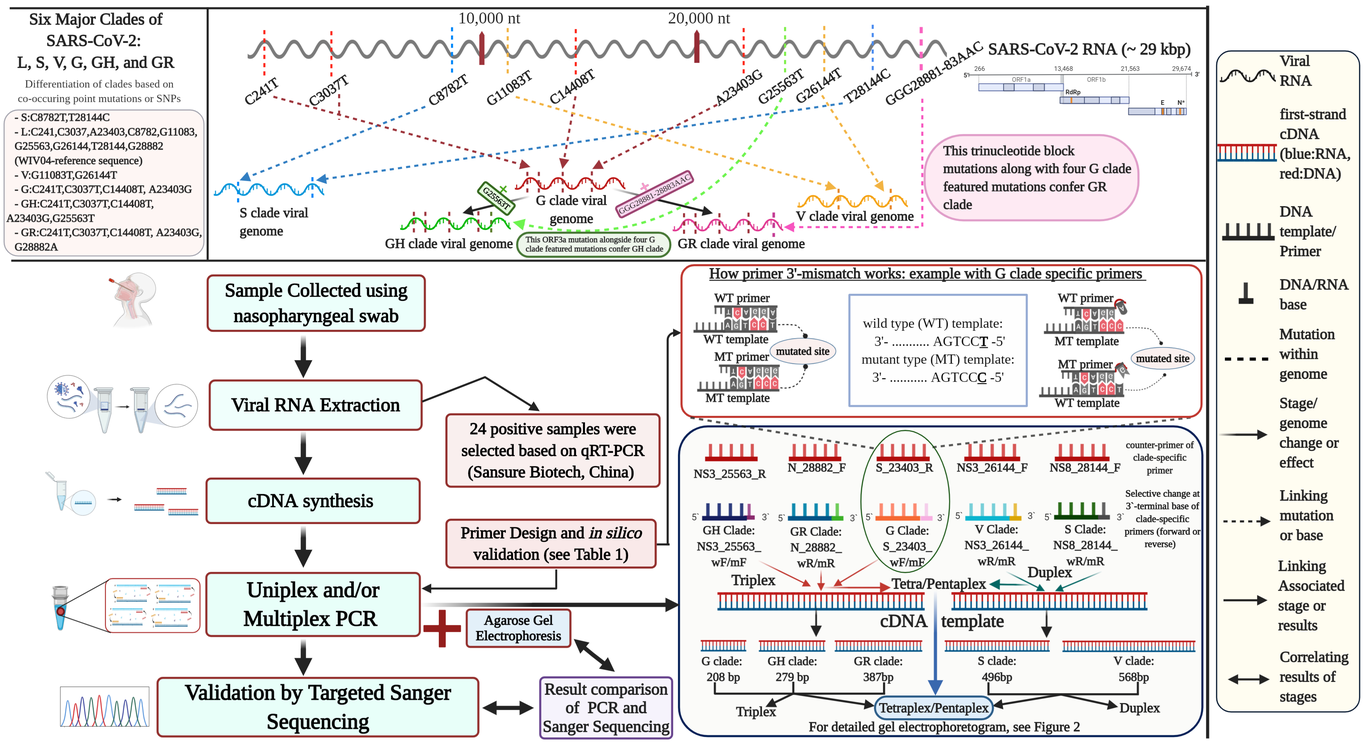
Tracing the globally circulating SARS-CoV-2 mutants is essential for the outbreak alerts and far-reaching epidemiological surveillance. The available technique to identify the phylogenetic clades through high-throughput sequencing is costly, time-consuming, and labor-intensive that hinders viral genotyping in low-income countries. Here, we propose a rapid, simple, and cost-effective amplification-refractory mutation system (ARMS)-based multiplex reverse-transcriptase PCR assay to identify six distinct phylogenetic clades: S, L, V, G, GH, and GR. This approach is applied on 24 COVID-19 positive samples as confirmed by CDC approved real-time PCR assay for SARS-CoV-2. Our multiplex PCR is designed in a mutually exclusive way to identify V-S and G-GH-GR clade variants separately. The pentaplex assay included all five variants and the quadruplex comprised of the triplex variants alongside either V or S clade mutations that created two separate subsets. The procedure was optimized in the primer concentration (0.2-0.6 µM) and annealing temperature (56-60°C) of PCR using a 3-5 ng/µl cDNA template synthesized upon random- and oligo(dT)-primer based reverse transcription. The different primer concentrations for the triplex and quadruplex adjusted to different strengths ensured an even amplification with a maximum resolution of all targeted amplicons. The targeted Sanger sequencing further confirmed the presence of the clade-featured mutations with another set of our designed primers. This multiplex ARMS-PCR assay is a sample, cost-effective, and convenient that can successfully discriminate against the circulating phylogenetic clades of SARS-CoV-2.