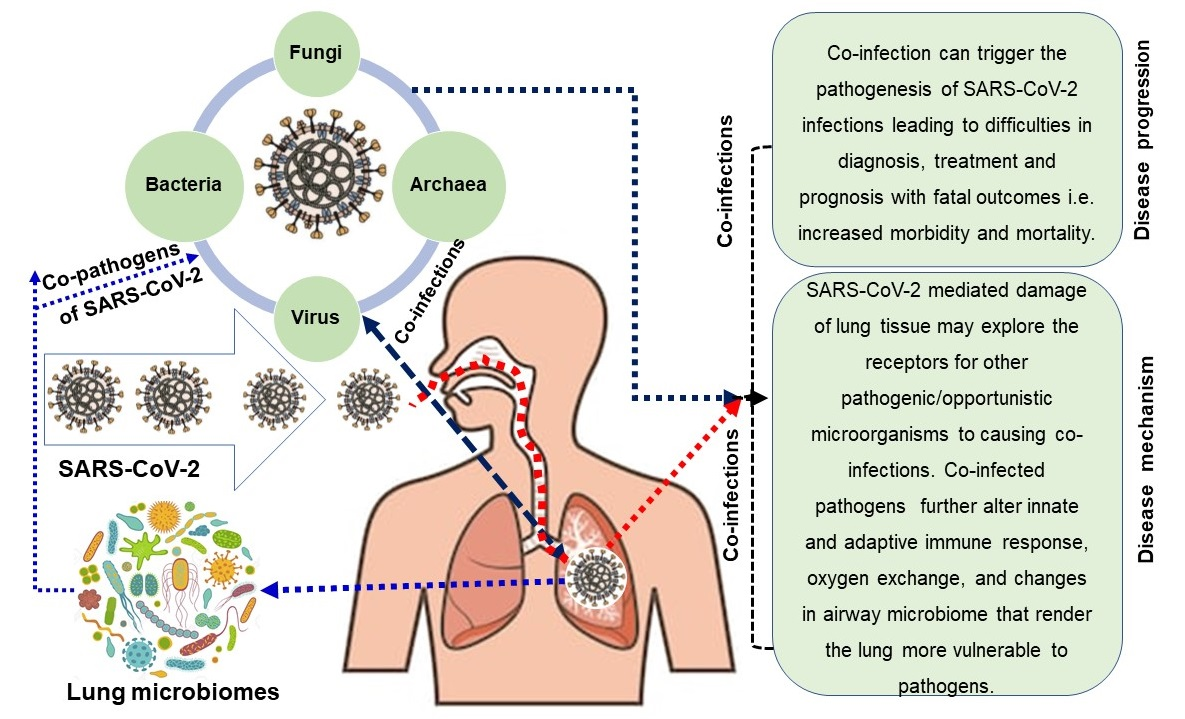
The emergence of novel coronavirus infectious disease-2019 (COVID-19) in December 2019, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has traumatized the whole world with the ongoing devastating pandemic. After droplet mediated transmission of infectious virus particle, and subsequent tissue tropism through the upper and lower respiratory tract, the acute clinical disease is manifested by severe respiratory illness accompanied by shortness of breath, progressive pneumonia, multi-organ dysfunction and ultimate death in SARS-CoV-2 infected patients. The involvement of other microbial co-infections leading to extortionate ailment in critically ill patients has not been significantly reviewed along with conclusive reporting on underlying molecular mechanisms in COVID-19 patients. Although the incidence of co-infections could be up to 94.2% in laboratory-confirmed COVID-19 cases, the fate of co-infections among SARS-CoV-2 infected hosts often depends on the balance between the host’s protective immunity and immunopathology. The cross-talk between co-pathogens (especially lung microbiomes), SARS-CoV-2 and host is an important factor that ultimately increases the difficulty of diagnosis, treatment, and prognosis of COVID-19, and even increase the symptoms and mortality of the disease. Simultaneously, co-infecting microorganisms may use new strategies to escape host defense mechanisms (by altering both innate and adaptive immune responses) to further aggravate SARS-CoV-2 pathogenesis. This review of literature suggests that clinicians should rule out SARS-CoV-2 infection by ruling in other respiratory co-pathogens, and must have a high index of suspicion for co-infection among COVID-19 patients. Thus, after recognizing the possible pathogens causing co-infection among COVID-19 patients, and the underlying molecular mechanisms of co-infections appropriate curative and preventive interventions can be recommended.