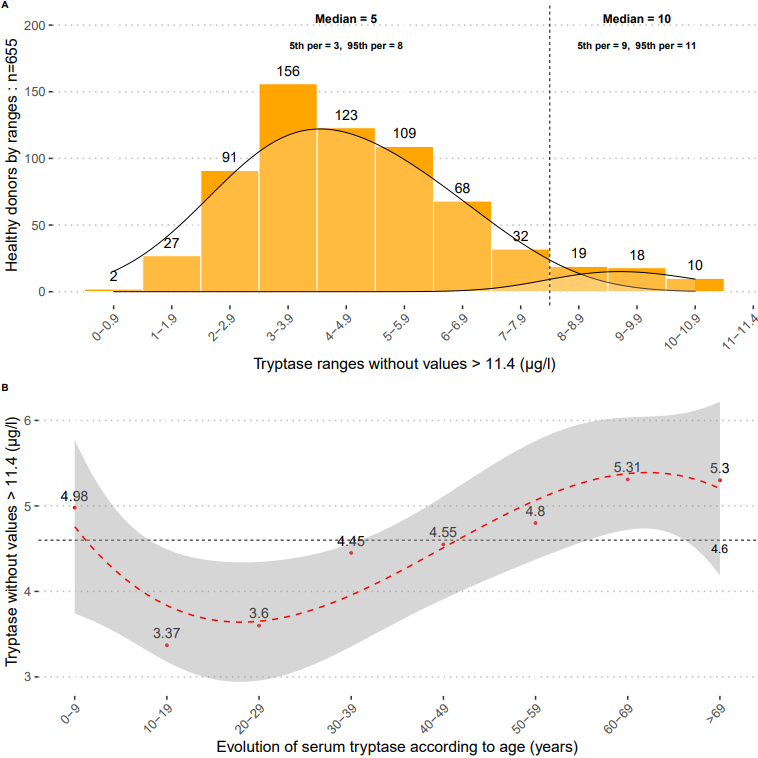
To the Editor, Precision medicine is increasingly used as an approach to the management of allergy and anaphylaxis, thanks to progress in diagnostic tests and biomarkers now allowing thorough characterization of a patient’s endotype1. Probability-based risk assessment and diagnostic algorithms have entered the allergists’ toolbox2-4. Allergy tests must therefore offer reliable, robust, and proficient results in each patient. Focusing onin vitro diagnostics, these requirements have led to the development of quality assurance (QA) programs for allergy laboratory assays and their implementation in virtually all clinical laboratories performing allergy assays. However, full performance targets for allergy assays have not yet been established, leaving allergists and clinical scientists without a common body of recommendations for the three routine assays, namely total serum IgE (tIgE), allergen-specific serum IgE (sIgE), and serum total tryptase. As an example, not only do recommendations on the acceptable bias and uncertainty of measurement (UM) of allergy assays miss from available literature, but there is also a complete lack of published recommendations on tryptase QA criteria. The multicentric French network of public clinical laboratories had previously documented a single-analyte QA strategy and recommendation for sIgE5. Hence, we set out to define QA criteria for intra- and interassay variation, analytical accuracy, and UM for sIgE, tryptase, and tIgE. QA data from 24 French centers were collected, analyzed, and compared to available literature, prior to issuing recommendations for QA management programs in allergy testing.Data were collected from 2016-2018 intralaboratory (internal) QA controls (IQA) and interlaboratory proficiency testing programs (external quality assurance, EQA) completed by the participant centers6. A literature search for English and French recommendations for allergy assays was performed, including scientific publications, statements of scientific societies, QA management schemes from independent QA organisms, and manufacturer documents. According to the regulated (tIgE) or nonregulated (sIgE, tryptase) analyte status7, the current work applies to any tIgE system, but for sIgE and total tryptase it is limited to the ImmunoCAP assay system, which is in use in all participant centers, is currently perceived as the reference in vitro diagnostic method for allergy2, and offers the only EU-cleared tryptase determination method. Briefly, IQA programs were performed with control samples provided by the manufacturer and with internal serum pools, particularly for tryptase determination. EQA programs were from UK NEQAS (UK National External Quality Assessment Services), Thermo Fisher Scientific (Uppsala, Sweden), ProBioQual (Lyon, France), and CTCB (Toulouse, France). All participant laboratories had subscribed to at least one EQA for each assay. Data analysis was performed stepwise: (1) definition of three concentration levels (low, medium, and high) within the dynamic range of each analyte and assignment of measurement results from each center to the corresponding level; (2) computation and analysis of intra- and interassay coefficient of variability (CV), bias from analytical accuracy, and UM for each analyte, concentration level, and participant; (3) comparison of assay performance of participant centers with extant recommendations, outlier identification and establishment of recommendations. Performance evaluation criteria were defined as follows: CV = 100xSD/mean (SD, standard deviation), bias = 100x[(participant result) – (peer group target result)]/(peer group target result), UM = √ [u2(IQA) + u2 (IQA)], with u2(IQA) denoting the variance (square SD) of all IQA results of the same concentration level, and u2(EQA) denoting the variance of corresponding EQA results8.Comparison of participant centers’ results and available recommendations (Table 1 ) revealed that actual tIgE assays outperformed most intra- and interassay CV recommendations, but were in line with bias recommendations. Actual sIgE assay performance for intra-and interassay CV matched the available non-manufacturer recommendations from CLSI (Clinical and Laboratory Standards Institute)9, but inconsistently attained UK NEQAS standards (Table 1 ). Intra-and interassay CV for total tryptase determination could only be compared to manufacturer recommendations, which appeared too stringent for inter-assay CV. Similarly, actual accuracy bias for tryptase determination was less performant than the available UK NEQAS standards, designed for low concentration levels (Table 1 ). For the three analytes and each concentration level, UM was calculated but due to a complete lack of available recommendations it could not be evaluated outside the peer group. Moreover, due to the lack of adequate EQA for each tryptase level, the UM for low (< 8 µg/L) and medium (8-20 µg/L) could only be computed for a combined low and medium concentration level up to 20 µg/L (Table 1 ).Analysis of data from participant centers and comparison with international standards (when available) allowed the establishment of recommended targets for performance evaluation, defined as the 95th percentile of the participants’ results (Table 2 ). It is noteworthy that UM, a performance criterion that should be considered whenever clinical interpretation and decision rely on quantitative results, needs improvement, both in terms of availability of adequate EQA samples spanning the whole range of analyte concentrations, and of results from participating centers. The first step to take is wider availability of IQA and EQA samples of paired concentration levels. As UM computation is based on the absolute value of variance, UM of low concentrations of an analyte is unfavorably impacted by the use of medium or high EQA sample results. In order to achieve the goal of using adequate pairs of EQA samples for each analyte level, in the absence of commercially available EQA programs, interlaboratory exchanges are a simple, cost-effective solution.In conclusion, we report here the first experience-based performance results for the most usual in vitro allergy and anaphylaxis assays, their comparison with available recommendations, and the establishment of the first recommendations for total tryptase assays and for the uncertainty of measurement of the three considered analytes: total serum IgE, allergen-specific serum IgE, and total serum tryptase. Conceived as a working tool for allergists and clinical scientists, our report aims at incentivizing further improvement and better use ofin vitro allergy assays for precision medicine.Anne Sarrat1, Rémy Couderc2, Marie-Alexandra Alyanakian3, Pol-André Apoil4, Céline Beauvillain5, Lionel Chollet6, Pascale Chrétien7, Arnaud Cirée8, Benoît Cypriani9, Erwan Dumontet10, Bertrand Evrard11, Lorna Garnier12, Angélique Grenier13, Valérie Guérin14, Caroline Hémont15, Anthony Léon16, Delphine Mariotte17, Pascale Nicaise-Roland18, Martine Pernollet19, Stéphanie Rogeau20, Thierry Tabary21, Béatrice Uring-Lambert22, Mylène Vivinus23, Julien Goret1, Joana Vitte24.1 Laboratoire d’Immunologie et Immunogénétique CHU Bordeaux, Hôpital Pellegrin, Bordeaux, France2 CHU Trousseau, Paris, France3 Laboratoire d’Immunologie, Hôpital Necker-Enfants Malades, AP-HP, Paris, France4 Institut Fédératif de Biologie, Hôpital Purpan, CHU Toulouse, Toulouse, France5 Laboratoire d’Immunologie, CHU Angers, France6 LBM CHI Toulon La Seyne sur Mer, Toulon, France7 Département d’Immunologie, AP-HP, Hôpitaux Universitaires Paris-Sud, Le Kremlin Bicêtre, France8 Laboratoire d’Immunologie, CHRU Tours, Tours, France9 Laboratoire de biochimie CHRU Besançon, Besançon, France10 CHU Rennes, Pôle Biologie, Rennes, France11 Service d’Immunologie, CHU Clermont-Ferrand, Clermont-Ferrand, France12 Laboratoire d’Immunologie, Hospices Civils de Lyon, Centre Hospitalier Lyon Sud, Pierre-Bénite, France13 LBM Hôpital Robert Ballanger, CHI Aulnay, France14 Laboratoire d’Immunologie, Hôpital Robert Debré, AP-HP, Paris, France15 Laboratoire d’immunologie, CHU Nantes, Nantes, France16 LBM CH Emile Durkheim, Epinal, France17 Département d’Immunologie et Immunopathologie, CHU Caen, Caen, France18 Laboratoire d’immunologie, « Autoimmunité et Hypersensibilités », Hôpital Bichat-Claude Bernard, AP-HP, Paris, France19 Institut de Biologie et de Pathologie, Laboratoire d’Immunologie, CHU Grenoble Alpes, Grenoble, France20 CHRU de Lille, Institut d’Immunologie-HLA, Lille, France21 Laboratoire d’immunologie, CHU Reims, Reims, France22 Département d’Immunobiologie, Hôpitaux Universitaires de Strasbourg, Strasbourg, France23 Laboratoire d’Immunologie, Hôpital de l’Archet, CHU Nice, France24 Aix Marseille Univ, IRD, University Hospitals of Marseille, MEPHI, Marseille, France