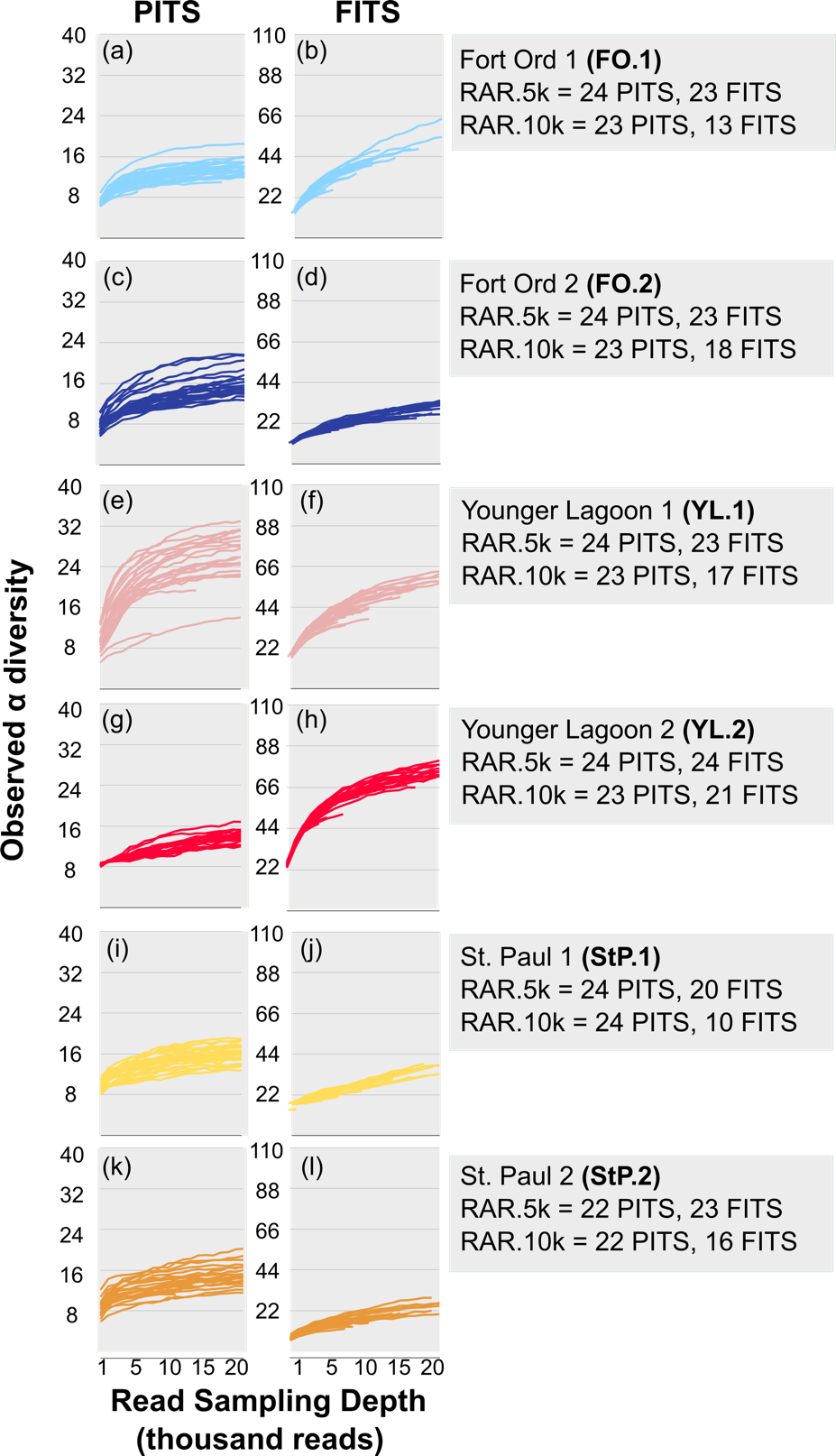
Environmental DNA (eDNA) metabarcoding is a common tool for measuring and cataloguing biodiversity, yet standard methodological approaches to generate metabarcoding data sets have yet to emerge, in part due to challenges understanding the biological and technical biases that affect eDNA profiles. Here, we explore how two experimental choices – depth of sequencing of PCR amplicon libraries and the number of PCR replicates – influence estimates of α and β diversity. We extracted DNA from six soil samples from three ecologically distinct locations, performed 24 PCR replicates from each using two common metabarcodes, and sequenced each to an average depth of 83,898 reads. We found PCR replicates are consistent in composition and relative abundance of abundant taxa, allowing differentiation of samples and sites. However, rare taxa were unique to one or a few replicates, suggesting that even large numbers of experimental replicates may be insufficient to catalogue biodiversity fully. We recommend that to differentiate sites, separately sequencing only a minimum of two PCR replicates to a depth that allows 1,000 reads identified to taxa, is sufficient to differentiate sites. We also conclude that metabarcoding is impractical for exhaustive taxonomic inventory and, because rare taxa are not amplified consistently, taxonomic tallies that rely on consensus among replicates artificially lower richness estimates. These findings provide new considerations for eDNA experimental design and data interpretation.