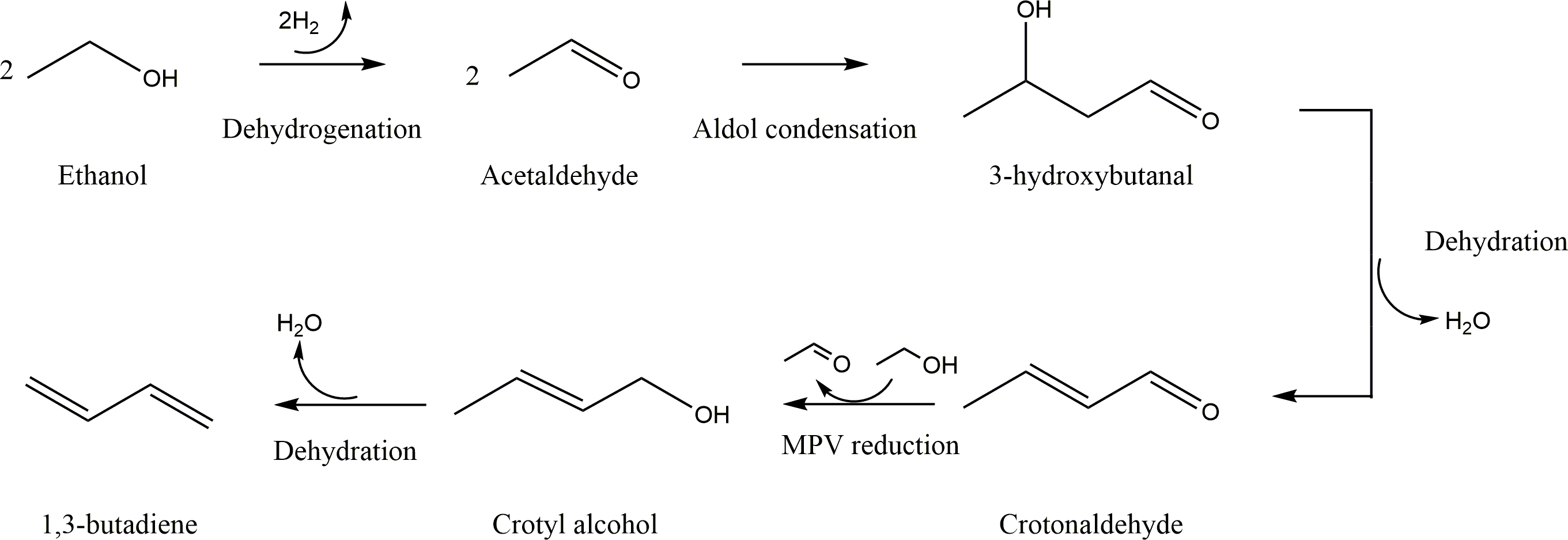
Density functional theory (DFT) calculations were conducted to investigate mechanistic details of ethanol-to-butadiene conversion reaction over MgO or ZnO catalyst. We evaluated the Lewis acidity and basicity of MgO and ZnO and found that ZnO had the stronger Lewis acidity and basicity compared with those of MgO. Potential energy surfaces (PESs) of ethanol-to-butadiene conversion, which included relevant transition states (TSs) and intermediates, were computed in detail following the generally accepted mechanism reported in the literature, where such mechanism included ethanol dehydrogenation, aldol condensation, Meerwein-Pondorf-Verley (MPV) reduction and crotyl alcohol dehydration. DFT results showed that ethanol dehydrogenation was the rate limiting step of overall reaction when the reaction was catalyzed by MgO. Also, DFT results showed that ethanol dehydrogenation occurred more easily on ZnO compared with MgO where such a result correlated with the stronger Lewis acidity of ZnO. In addition, we computed ethanol dehydration which generates ethylene, one of the major undesired side reaction products for butadiene formation. DFT results showed that ZnO favored dehydrogenation over dehydration while MgO favored dehydration.