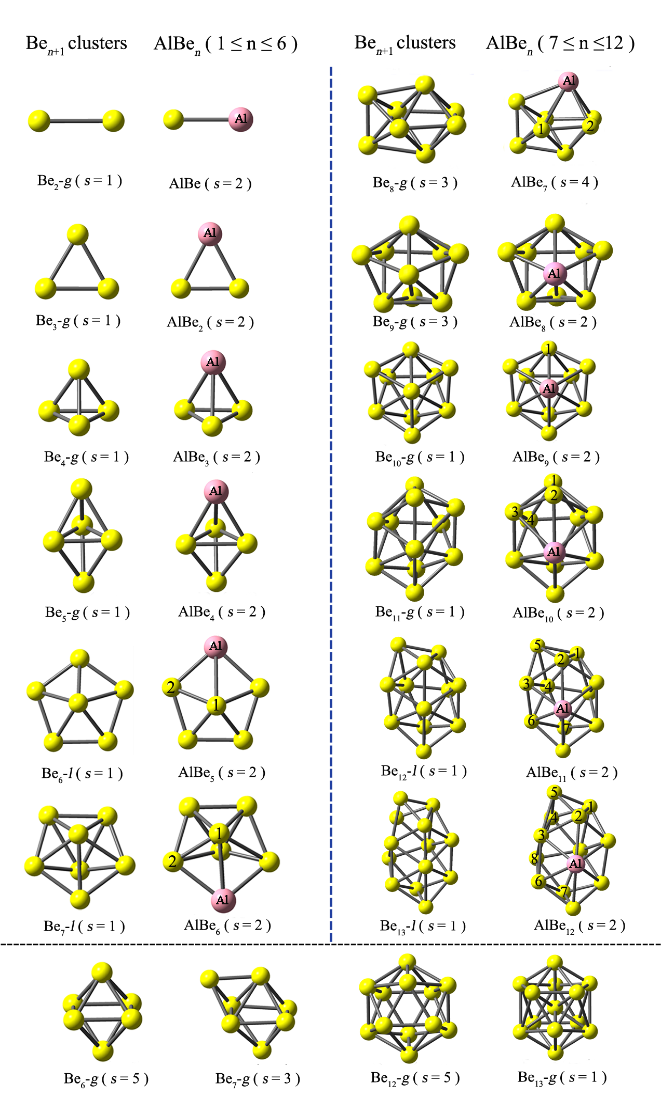
The geometric structures, energetic and electronic properties of global minima of the AlBen (n = 1–12) clusters have been systemically studied by using the hybrid density functional theory [B3LYP] and coupled cluster [CCSD(T)] methods. It is found that the impurity Al atom is externally bound to the host Ben framework and its maximum coordination number is six. Besides, the geometries of AlBen bear close resemblance to either local or global minimum structures of Ben+1. The AlBe3 and AlBe8 clusters exhibit high relative stability among the AlBen clusters, which is reflected by the evolutions of average atomic binding energy, dissociation energy, second difference in energy, adsorption energy of Al, and HOMO-LUMO gap with cluster size. In comparison to the pure Ben+1 clusters, AlBen exhibit larger binding energy values, whereas they are more polarizable.