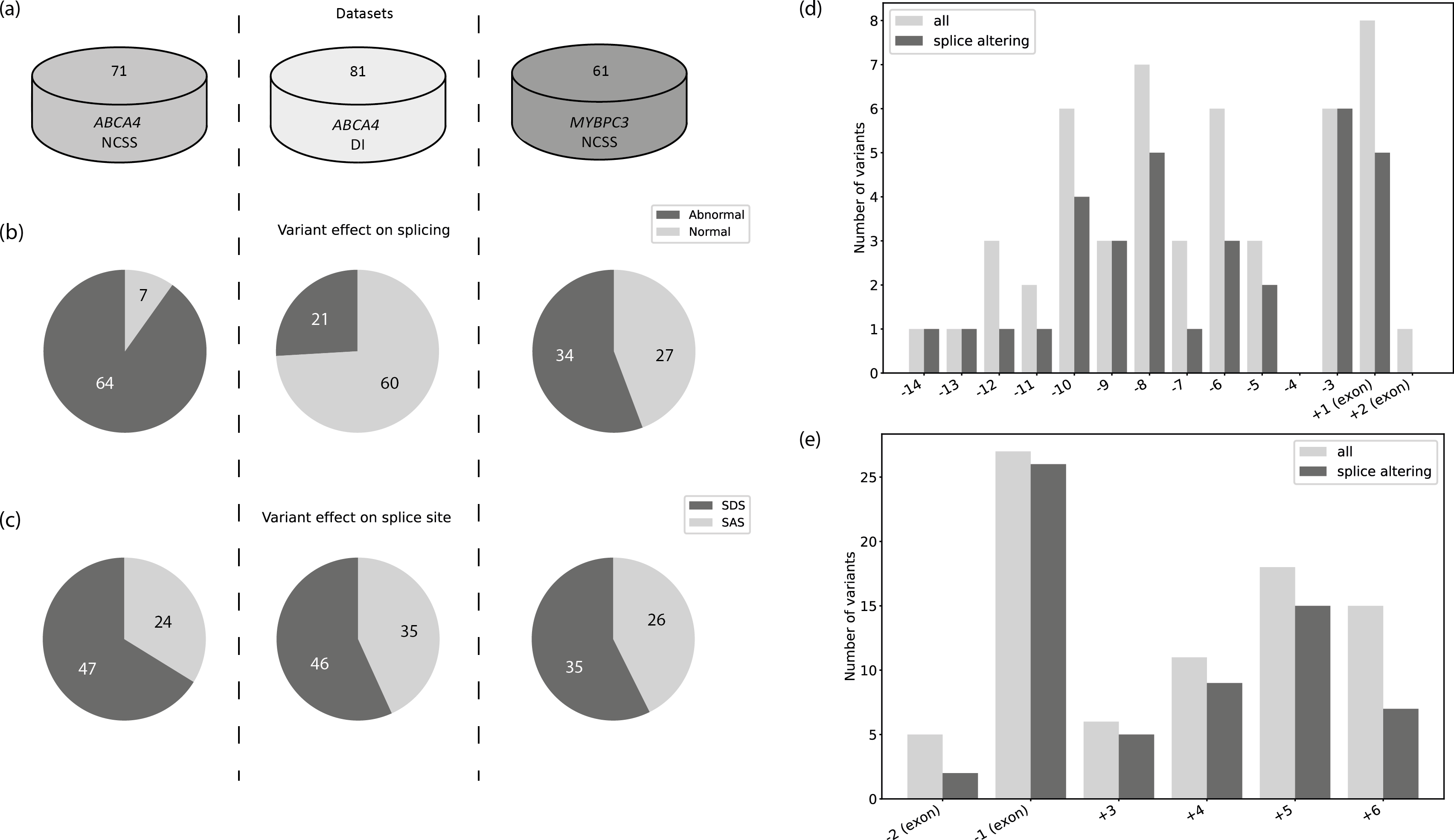
Hereditary disorders are frequently caused by genetic variants that affect pre-mRNA splicing. Whilst genetic variants in the canonical splice motifs are almost always disrupting splicing, the pathogenicity of variants in the non-canonical splice sites (NCSS) and deep intronic (DI) regions are difficult to predict. Multiple splice prediction tools have been developed for this purpose, with the latest tools employing deep learning algorithms. We benchmarked established and deep learning splice prediction tools on gold standard sets of variants in the ABCA4 and MYBPC3 genes associated with Stargardt disease (STGD1) and cardiomyopathy, respectively, with functional assessment in midigene and minigene splice assays. The best performing splice prediction tool for both NCSS and DI variants in ABCA4 was SpliceAI, whilst SpliceSiteFinder-like performed best for NCSS variants in MYBPC3. Overall, the performance in a real time clinical setting is much more modest than reported by the developers of the tools.