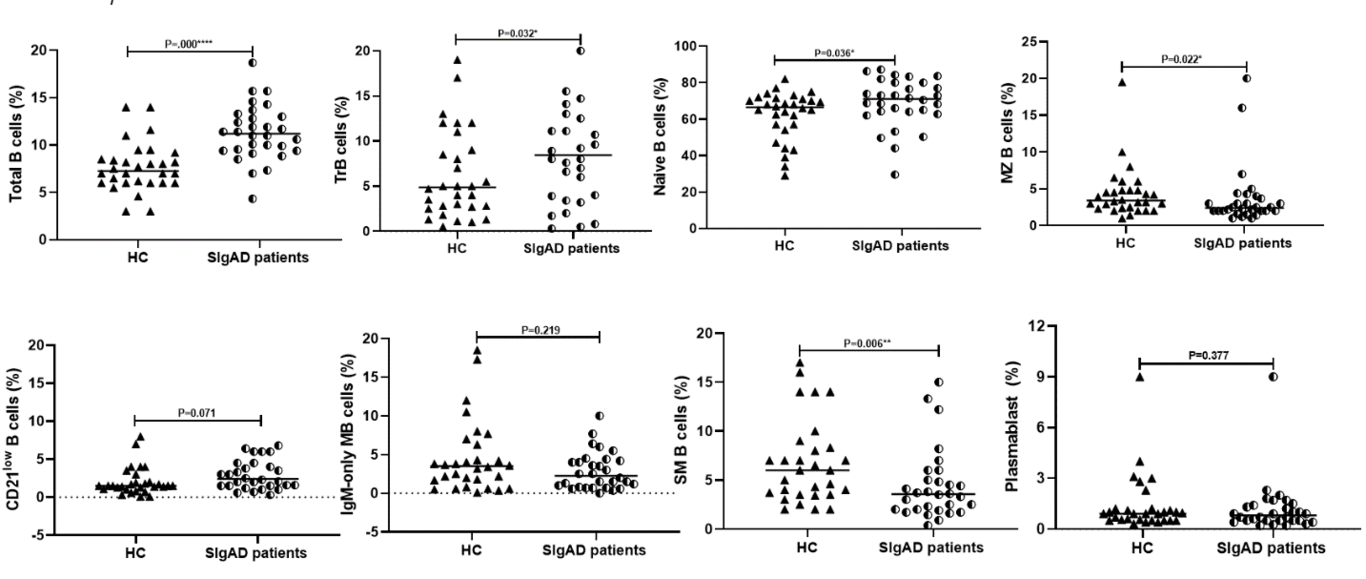
Background: Investigating and documenting novel mutations within inborn errors of immunity (IEIs) has demonstrably enriched our comprehension of disease causation. This encompasses elucidating the clinical and immunological presentations linked to specific gene alterations, ultimately leading to refined classification precision and, optimistically, improved treatment modalities. The present study aims to the profiles of patients within the Iranian IEI registry exhibiting mutations within the NFKB signaling pathway. Methods: Peripheral blood mononuclear cells were utilized for immunophenotyping of B and T lymphocyte subsets, and proliferation assays. Immunoblotting was employed to assess the expression levels of the corresponding protein in each patient harboring the respective variant. Results: The study cohort encompassed 17 patients: 8 with NFKB1 mutations, 5 with NFKB2 mutations, 3 with IKBKB mutations, and 1 with an IKBKG mutation. All NFKB1 and NFKB2 mutations presented as heterozygous, whereas IKBKB mutations were homozygous and the IKBKG mutation was hemizygous. The predominant clinical features included hypogammaglobulinemia and B cell subset disturbances, with T cell subsets and proliferation being normal in most, though not all, cases. Protein expression in patients generally mirrored healthy controls, except for two individuals harboring NFKB2 mutations. Conclusions: These findings provide novel IEI cases linked to NFKB pathway mutations. Comprehensive evaluation and functional analysis of novel mutations, confirming potential impacts on disease manifestation, also highlight the need for specialized care and further research within immunodeficiency disorders.