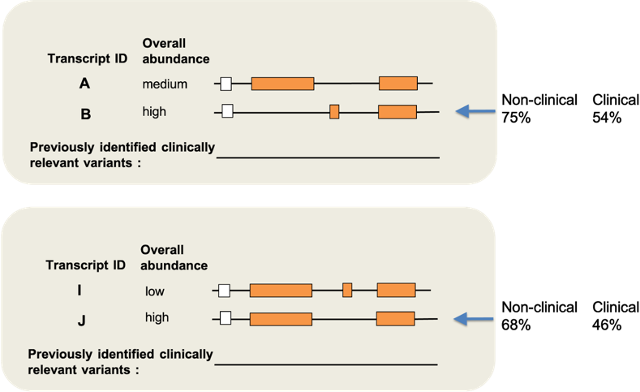
Variant interpretation is dependent on transcript annotation and remains time consuming and challenging. There are major obstacles for historical data reuse and for interpretation of new variants. First, both RefSeq and Ensembl/GENCODE produce transcript sets in common use, but there is currently no easy way to translate between the two. Second, the resources often used for variant interpretation (e.g. ClinVar, gnomAD, UniProt) do not use the same transcript set, nor default transcript or protein sequence. Ensembl ran a survey in 2018 to assay attitudes to choosing one default transcript per locus, and to gather data on reference sequences used by the scientific community. This was publicised on the Ensembl and UCSC genome browsers, by email and on social media. We had 788 respondents. Here we report our results and roadmap to create an effective default set of transcripts for resources, and for reporting interpretation of clinical variants.