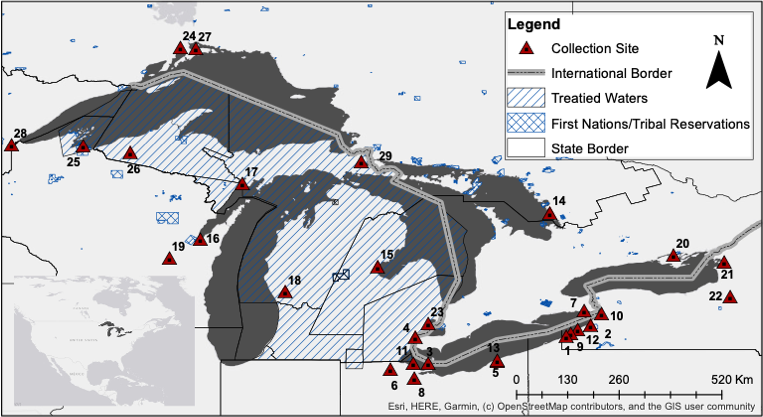
Artificial propagation and wild release may influence the genetic integrity of wild populations. This practice has been prevalent in fisheries for millennia and is often termed “stocking”. In the Laurentian Great Lakes, walleye populations faced declines from the 1950s to the 1970s, prompting extensive stocking efforts for restoration. By the mid-2010s, walleye populations showed signs of recovery, but the genetic legacy of stocking on population structure at the genomic level remains unclear. Using a dataset of 45,600 genome-aligned SNP loci genotyped in 1,075 walleye individuals, we investigated the genetic impacts of over 50 years of stocking across the Great Lakes. Natural geographic barriers shaped walleye population structure, but pairwise comparisons revealed changes in genetic structure due to stocking from non-native sources also significantly contribute to population structure. Admixture between Lake Erie walleye and walleye from the re-populated Tittabawassee River indicate that stocking may have re-distributed putatively adaptive alleles around the Great Lakes. Genome scans identified FST outliers and evidence of selective sweeps, indicating local adaptation of spawning populations is likely. Notably, one genomic region showed strong differentiation between Muskegon River and walleye from the Tittabawassee River which was re-populated by Muskegon Strain walleye, suggesting admixture and selection both impact the observed genetic diversity. Overall, our study underscores how artificial propagation and translocations can significantly alter the evolutionary trajectory of populations. The findings highlight the complex interplay between stocking practices and genetic diversity, emphasizing the need for careful management strategies to preserve the genetic integrity of wild populations amidst conservation efforts.