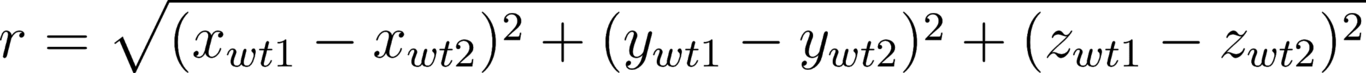When two or more amino acid mutations occur in protein systems, they can interact in a non-additive fashion termed epistasis. One way to quantify epistasis between mutation pairs in protein systems is by using free energy differences: ϵ = 𝚫𝚫G1,2 - (𝚫𝚫G1 + 𝚫𝚫G2) where 𝚫𝚫G refers to the change in the Gibbs free energy, subscripts 1 and 2 refer to single mutations in arbitrary order and 1,2 refers to the double mutant. In this study, we explore possible biophysical mechanisms that drive pairwise epistasis in both protein-protein binding affinity and protein folding stability. Using the largest available datasets containing experimental protein structures and free energy data, we derived statistical models for both binding and folding epistasis (ϵ) with similar explanatory power (R2) of 0.299 and 0.258, respectively. These models contain terms and interactions that are consistent with intuition. For example, increasing the Cartesian separation between mutation sites leads to a decrease in observed epistasis for both folding and binding. Our results provide insight into factors that contribute to pairwise epistasis in protein systems and their importance in explaining epistasis. However, the low explanatory power indicates that more study is needed to fully understand this phenomenon.