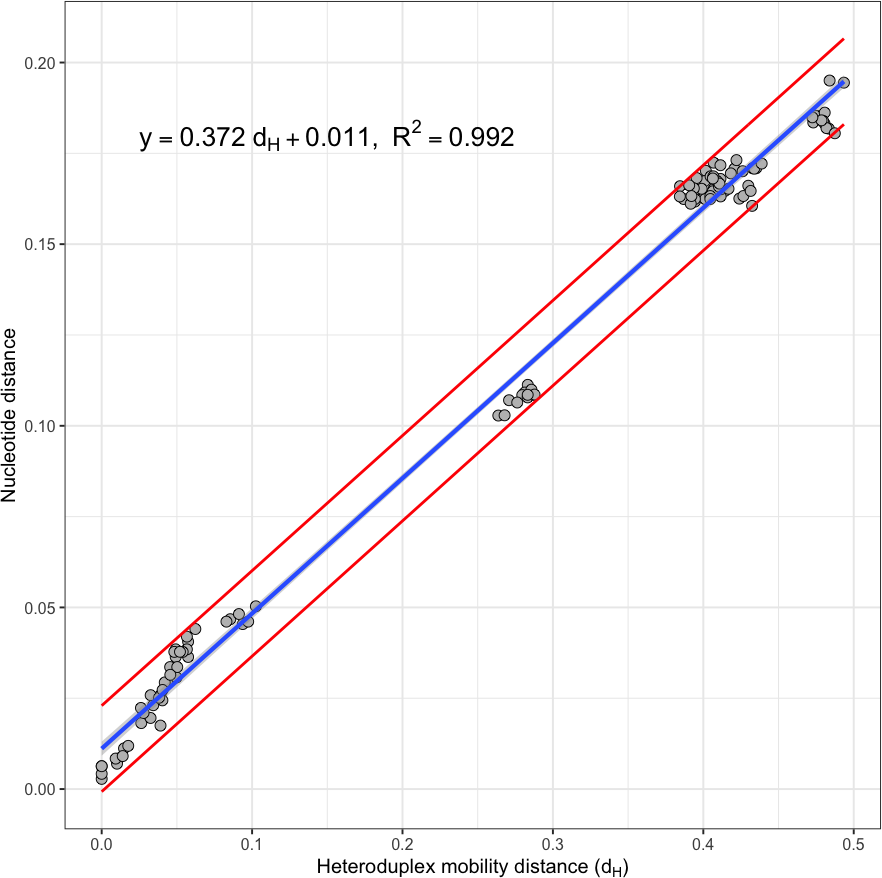
The Heteroduplex mobility assay (HMA) has proven to be a robust tool for the detection of genetic variation. Here, we describe a simple and rapid application of the HMA by microfluidic capillary electrophoresis, for phylogenetics and population genetic analyses (pgHMA). We show how commonly applied techniques in phylogenetics and population genetics have equivalents with pgHMA: phylogenetic reconstruction with bootstrapping, skyline plots, and mismatch distribution analysis. We assess the performance and accuracy of pgHMA by comparing the results obtained against those obtained using standard methods of analyses applied to sequencing data. The resulting comparisons demonstrate that: (1) there is a significant linear relationship (R = 0.992) between heteroduplex mobility and genetic distance; (2) phylogenetic trees obtained by HMA and nucleotide sequences present nearly identical topologies; (3) clades with high pgHMA parametric bootstrap support also have high bootstrap support on nucleotide phylogenies; (4) skyline plots estimated from the UPGMA trees of HMA and Bayesian trees of nucleotide data reveal similar trends, especially for the median trend estimate of effective population size; and (5) optimized mismatch distributions of HMA are closely fitted to the mismatch distributions of nucleotide sequences. In summary, pgHMA is an easily-applied method for approximating phylogenetic diversity and population trends. KEYWORDS: bootstrap, heteroduplex mobility assay, mismatch distribution, phylogenetics, skyline plot