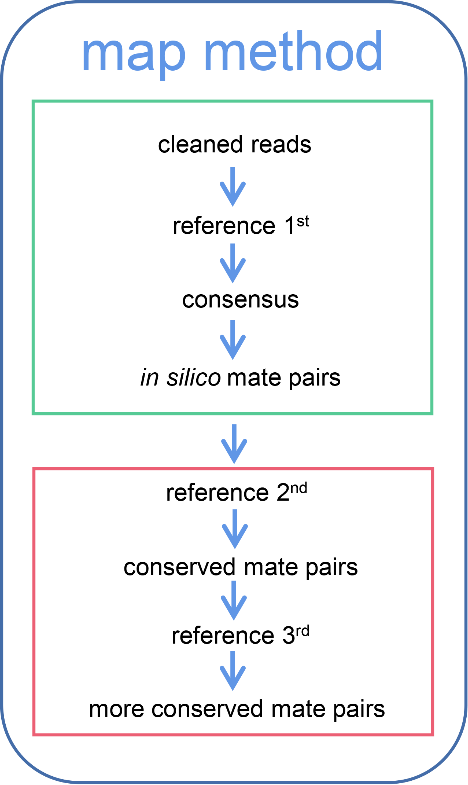
The elongate ilisha (Ilisha elongata) is a significant commercial species found along the Northwestern Pacific Coast. A sharp decline in the annual catch of I. elongata over recent decades implies a concerning situation regarding its fishery stocks. Nonetheless, inadequate knowledge of the genetic diversity, population structure, and historical demography of this species has hindered the establishment of sustainable fishery policies and appropriate conservation measures. In this study, the genetic structure and population demography of I. elongata stocks along the Northwestern Pacific Coast were examined using target-gene enrichment data from 144 I. elongata individuals collected from 18 locations. The findings reveal that with an average value of 0.2173, I. elongata maintains a notable level of nucleotide diversity despite facing considerable fishing pressure. Furthermore, inter-population differentiation is relatively low, with most geographical populations displaying minimal genetic distinctions or none from one another. Population clustering analysis identified four lineages of I. elongata stocks. Through historical demography simulations, it was proposed that the Yalu River Estuary population diverged initially around 19500 generations before present, while the remaining lineage split into two about 18800 generations ago. One lineage represents the southern population, while the other further separated into the northern population and the Japanese population approximately 3000 generations ago. These results underscore that the current phylogeographic patterns of I. elongata may result from directional selection due to low temperatures and geographic barriers during glacial periods, followed by recent expansions.