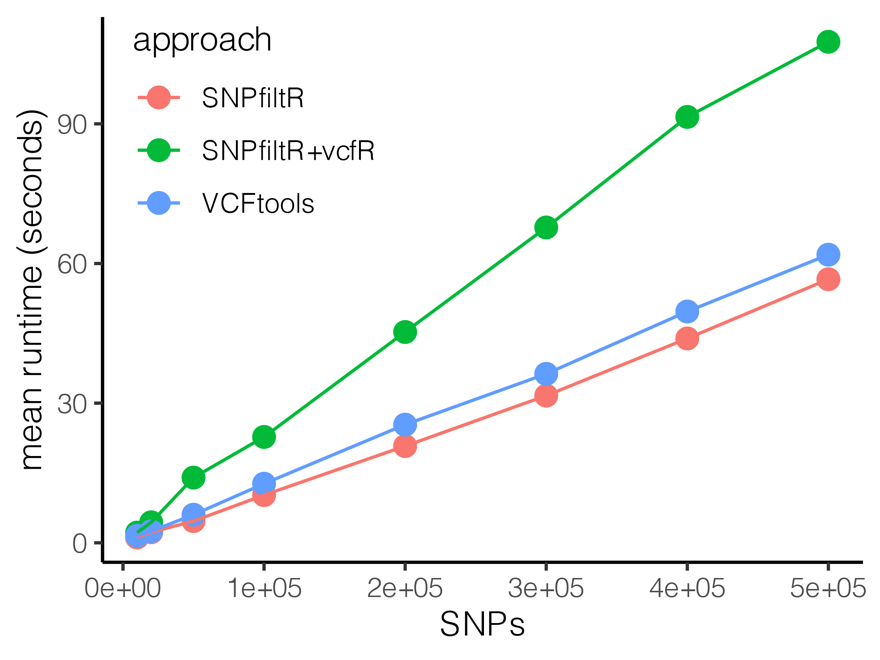
Here I describe the novel R package SNPfiltR and demonstrate its functionalities as the backbone of a customizable, reproducible SNP filtering pipeline implemented exclusively via the widely adopted R programming language. SNPfiltR extends existing SNP filtering functionalities by automating the visualization of key parameters such as depth, quality, and missing data, then allowing users to set filters based on optimized thresholds, all within a single, cohesive working environment. All SNPfiltR functions require a vcfR object as input, which can be easily generated by reading a SNP dataset stored as a standard vcf file into an R working environment using the function read.vcfR() from the R package vcfR. Performance benchmarking reveals that for moderately sized SNP datasets (up to 50M genotypes with associated quality information), SNPfiltR performs filtering with comparable efficiency to current state of the art command-line-based programs. These benchmarking results indicate that for most reduced-representation genomic datasets, SNPfiltR is an ideal choice for investigating, visualizing, and filtering SNPs as part of a cohesive and easily documentable bioinformatic pipeline. The SNPfiltR package can be downloaded from CRAN with the command [install.packages(“SNPfiltR”)], and a development version is available from GitHub at: (github.com/DevonDeRaad/SNPfiltR). Additionally, thorough documentation for SNPfiltR, including multiple comprehensive vignettes, is available at the website: (devonderaad.github.io/SNPfiltR/).