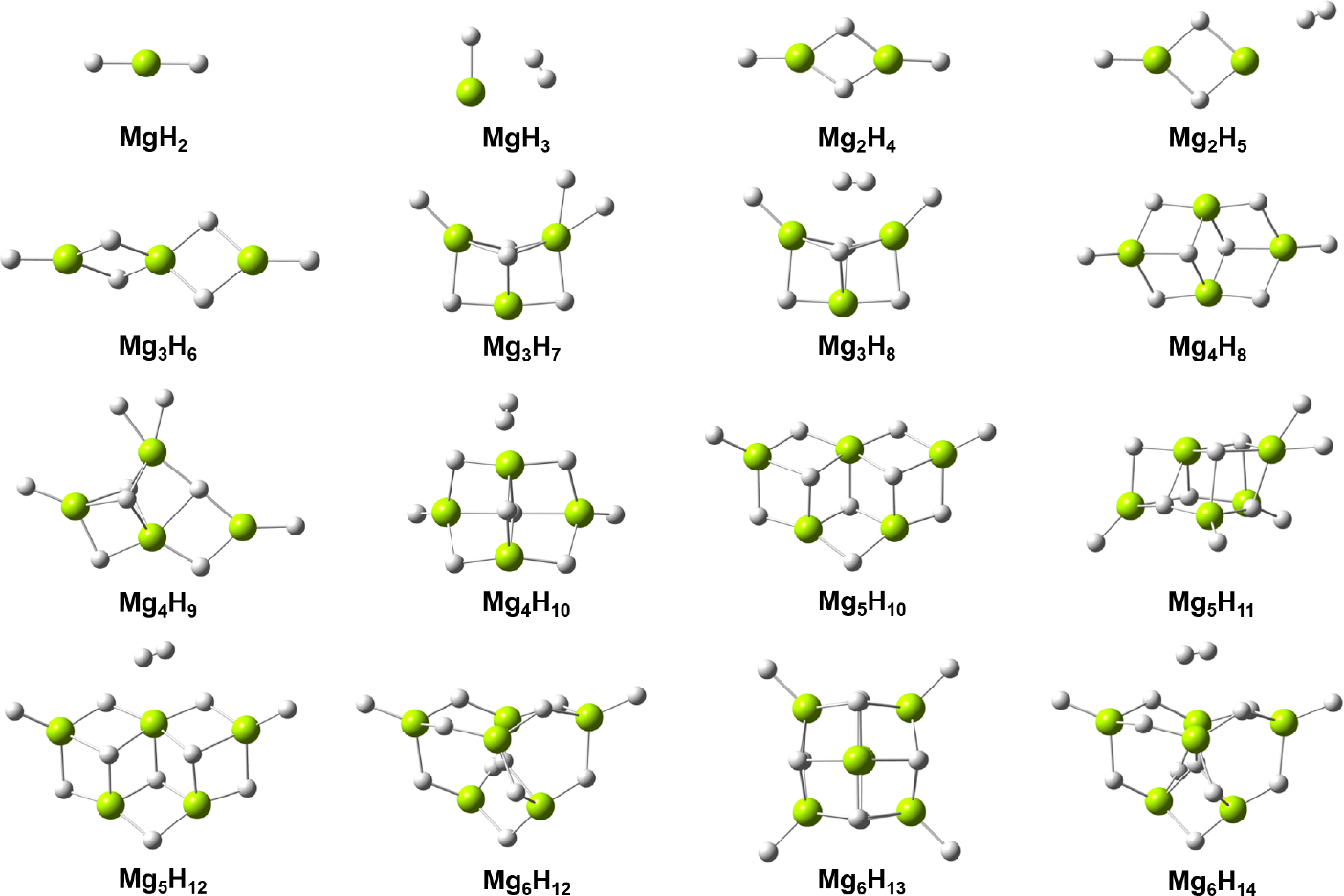
Magnesium-based hydrogen storage material (MgH2) has attracted much attention due to its high hydrogen storage density (7.6 wt%). However, the high hydrogen dissociation enthalpy and slow hydrogen dissociation rate in bulk Mg hinder its wide application in the efficient hydrogen storage. In the present work, we study the hydrogen adsorption and desorption reactions of MgmHn (m = 1-6) nanoclusters using density functional theory (DFT). From the global search for the configurations of MgmHn nanoclusters, we found not only stable saturated MgmHn (n = 2m) nanoclusters, but four hydrogen-enriched MgmHn (n:m>2:1) nanoclusters, Mg3H7, Mg4H9, Mg5H11, Mg6H13, with the hydrogen storage density higher than 8.3 wt%. The electronic-structure calculations indicate that the stability of the hydrogen-enriched cluster gets relatively higher for larger nanocluster. The ab initio dynamics simulations shows that all hydrogen-enriched clusters have very fast hydrogen dissociation rates, which is promising for the hydrogen dissociation at ambient temperature and pressure. This work provides insights into the hydrogen storage mechanism of nano-magnesium materials.