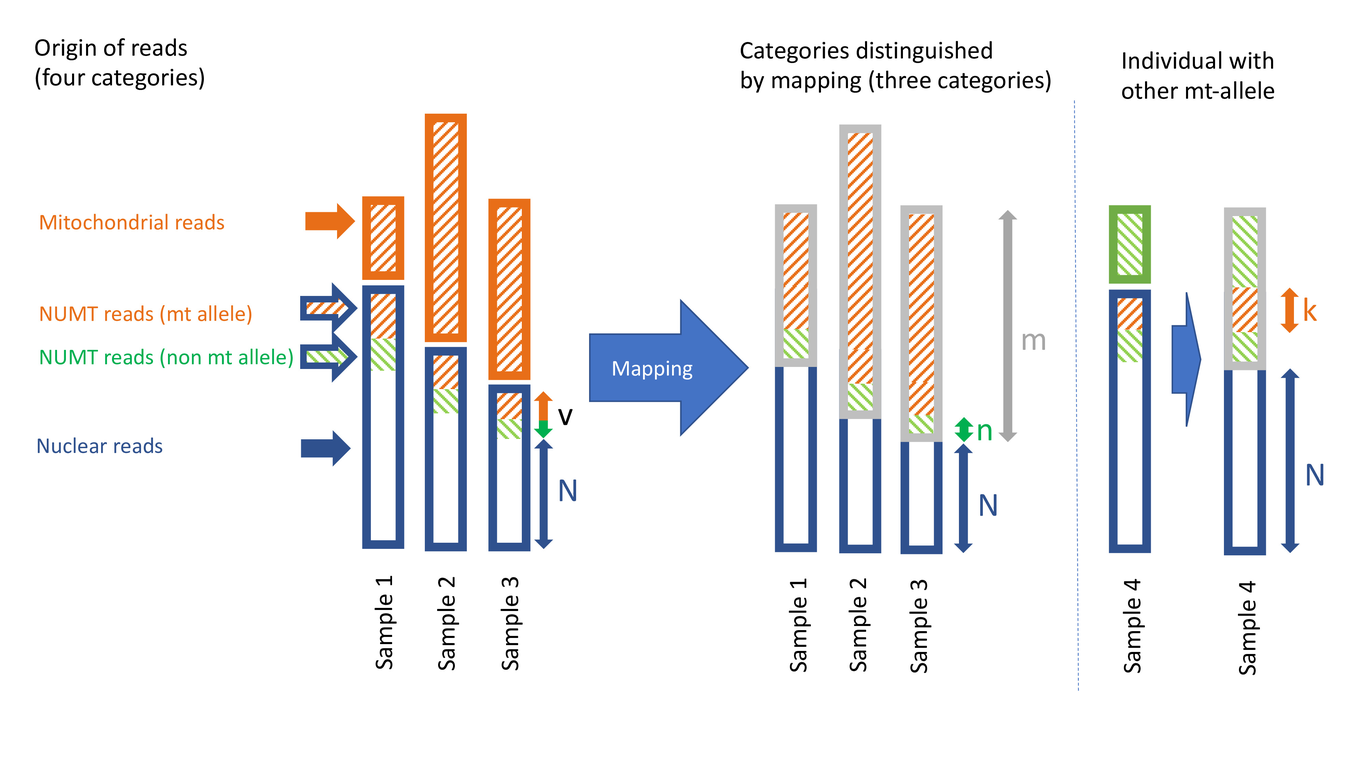
Inserts of DNA from extranuclear sources, such as organelles and microbes, are common in eukaryote nuclear genomes. However, sequence similarity between the nuclear and extranuclear DNA, and a history of multiple insertions, make the assembly of these regions challenging. Consequently, the number, sequence, and location of these vagrant DNAs cannot be reliably inferred from the genome assemblies of most organisms. We introduce two statistical methods to estimate the abundance of nuclear inserts even in the absence of a nuclear genome assembly. The first (intercept method) only requires low-coverage (<1x) sequencing data, as commonly generated for population studies of organellar and ribosomal DNAs. The second method additionally requires that a subset of the individuals carry extra-nuclear DNA with diverged genotypes. We validated our intercept method using simulations and by re-estimating the frequency of human NUMTs (nuclear mitochondrial inserts). We then applied it to the grasshopper Podisma pedestris, exceptional for both its large genome size and reports of numerous NUMT inserts, estimating that NUMTs make up 0.056% of the nuclear genome, equivalent to >500 times the mitochondrial genome size. We also re-analysed a museomics dataset of the parrot Psephotellus varius, obtaining an estimate of only 0.0043%, in line with reports from other species of bird. Our study demonstrates the utility of low-coverage high-throughput sequencing data for the quantification of nuclear vagrant DNAs. Beyond quantifying organellar inserts, these methods could also be used on endosymbiont-derived sequences. We provide an R implementation of our methods called “vagrantDNA” and code to simulate test datasets.