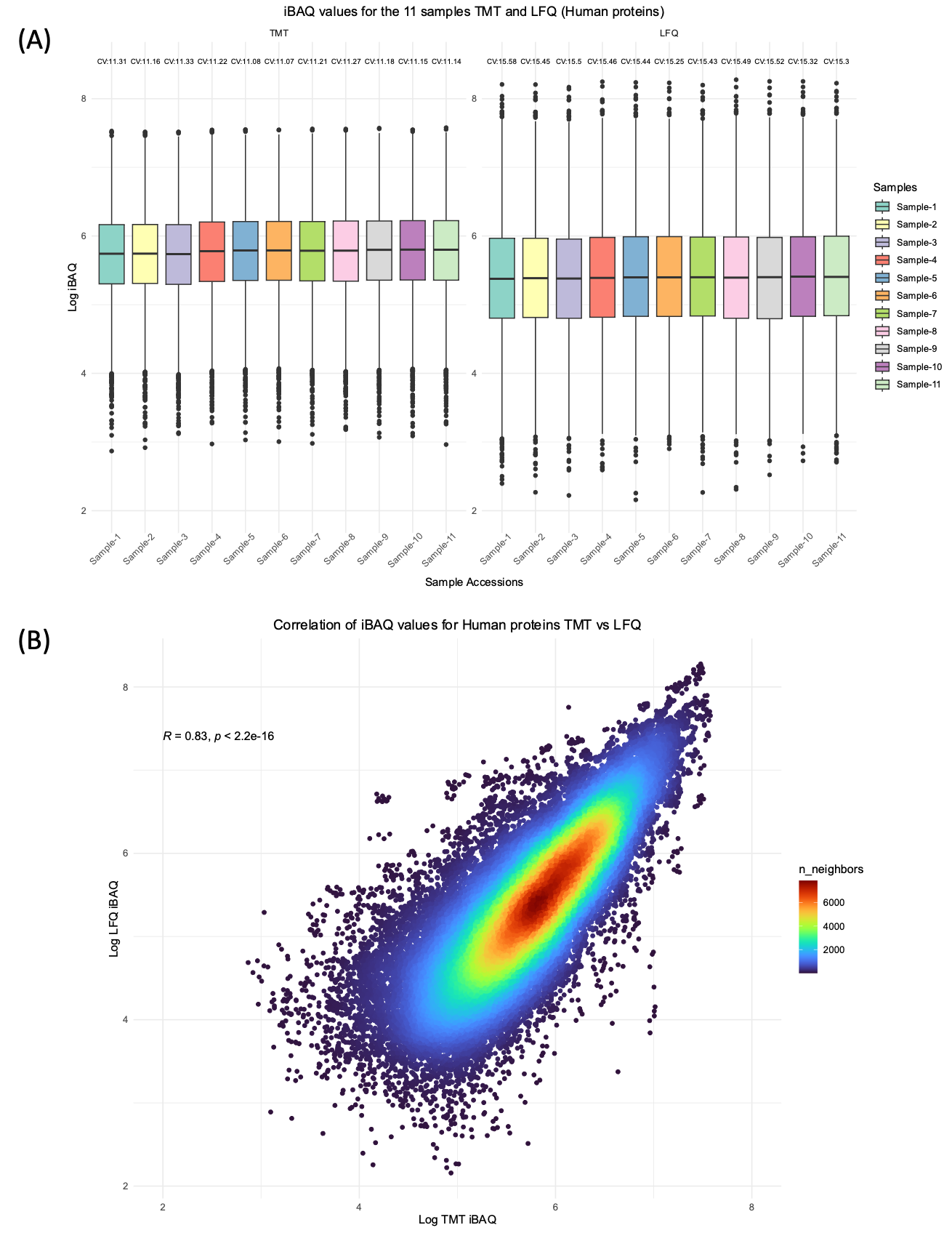
Relative and absolute intensity-based protein quantification across cell lines, tissue atlases, and tumour datasets is increasingly available in public datasets. These atlases enable researchers to explore fundamental biological questions, such as protein existence, expression location, quantity, and correlation with RNA expression. Most studies provide MS1 feature-based label-free quantitative (LFQ) datasets; however, growing numbers of isobaric tandem mass tags (TMT) datasets remain unexplored. Here, we compare traditional intensity-based absolute quantification (iBAQ) proteome abundance ranking to an analogous method using reporter ion proteome abundance ranking with data from an experiment where LFQ and TMT were measured on the same samples. This new TMT method substitutes reporter ion intensities for MS1 feature intensities in the iBAQ framework. Additionally, we compared LFQ-iBAQ values to TMT-iBAQ values from two independent large-scale tissue atlas datasets (one LFQ and one TMT) using robust bottom-up proteomic identification, normalisation, and quantitation workflows.