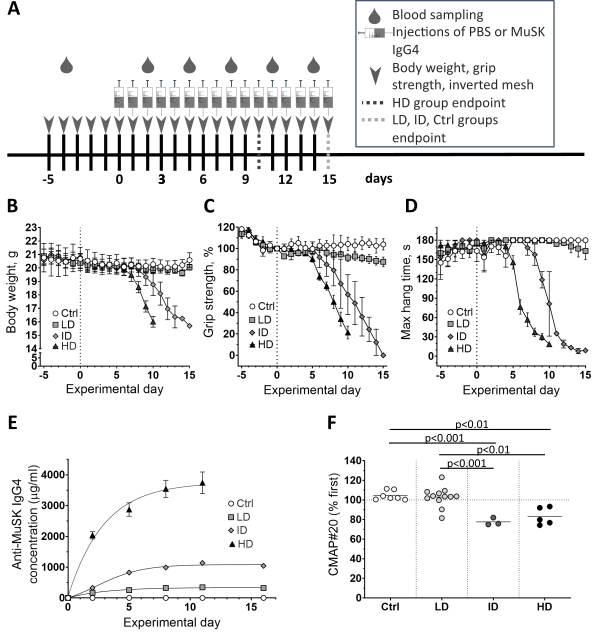
Muscle-specific kinase myasthenia gravis (MuSK MG) is caused by autoantibodies against MuSK in the neuromuscular junction (NMJ). MuSK MG patients have fluctuating, fatigable weakness, in particular of bulbar muscles. Severity differs greatly between patients, in spite of comparable autoantibody levels. One explanation for inter-patient and inter-muscle variability in sensitivity might be variations in compensatory muscle responses. Previously, we developed a passive transfer mouse model for MuSK MG. In preliminary ex vivo experiments we observed that muscle contraction, in particular of mice with milder myasthenia, had become partially insensitive to μ-Conotoxin-GIIIB, a blocker of skeletal muscle NaV1.4 voltage-gated sodium channels. We hypothesized that changes in NaV channel expression profile, possibly co-expression of (μ-Conotoxin-GIIIB insensitive) NaV1.5 type channels, might lower the muscle fibre’s firing threshold and facilitate neuromuscular synaptic transmission. To test this, we here performed passive transfer in mice, using ‘high’, ‘intermediate’ and ‘low’ dosing regimens of purified MuSK MG patient IgG4 and compared myasthenia levels, μ-Conotoxin-GIIIB resistance, muscle fibre action potential characteristics and firing thresholds. High- and intermediate-dosed mice showed clear, progressive myasthenia, not seen in low-dosed animals. However, diaphragm NMJ electrophysiology demonstrated almost equal myasthenic severities amongst all regimens. Nonetheless, low-dosed mouse diaphragms showed a much higher degree of μ-Conotoxin-GIIIB resistance. This was not explained by upregulation of Scn5a (the NaV1.5 gene), lowered muscle fibre firing thresholds or histologically detectable upregulated NaV1.5 channels. It remains to be established which factors are responsible for the μ-Conotoxin-GIIIB insensitivity and whether the NaV repertoire change is compensatory beneficial, or a bystander effect.