AUTHOREA
Log in
Sign Up
Browse Preprints
LOG IN
SIGN UP
Submit
Join
Quantum Simulations Group, Lawrence Livermore National Laboratory, Livermore, California 94550, USA
1,289
views
35
downloads
Editor:
Xavier Andrade
Public Documents
2
Members
by author
by title
by keyword
Filter
All
All
Version of Record
Sort by
Most Recent
Most Recent
Most Viewed
Most Cited
Real-space grids and the Octopus code as tools for the development of new simulation...
Xavier Andrade
and 15 more
August 18, 2014
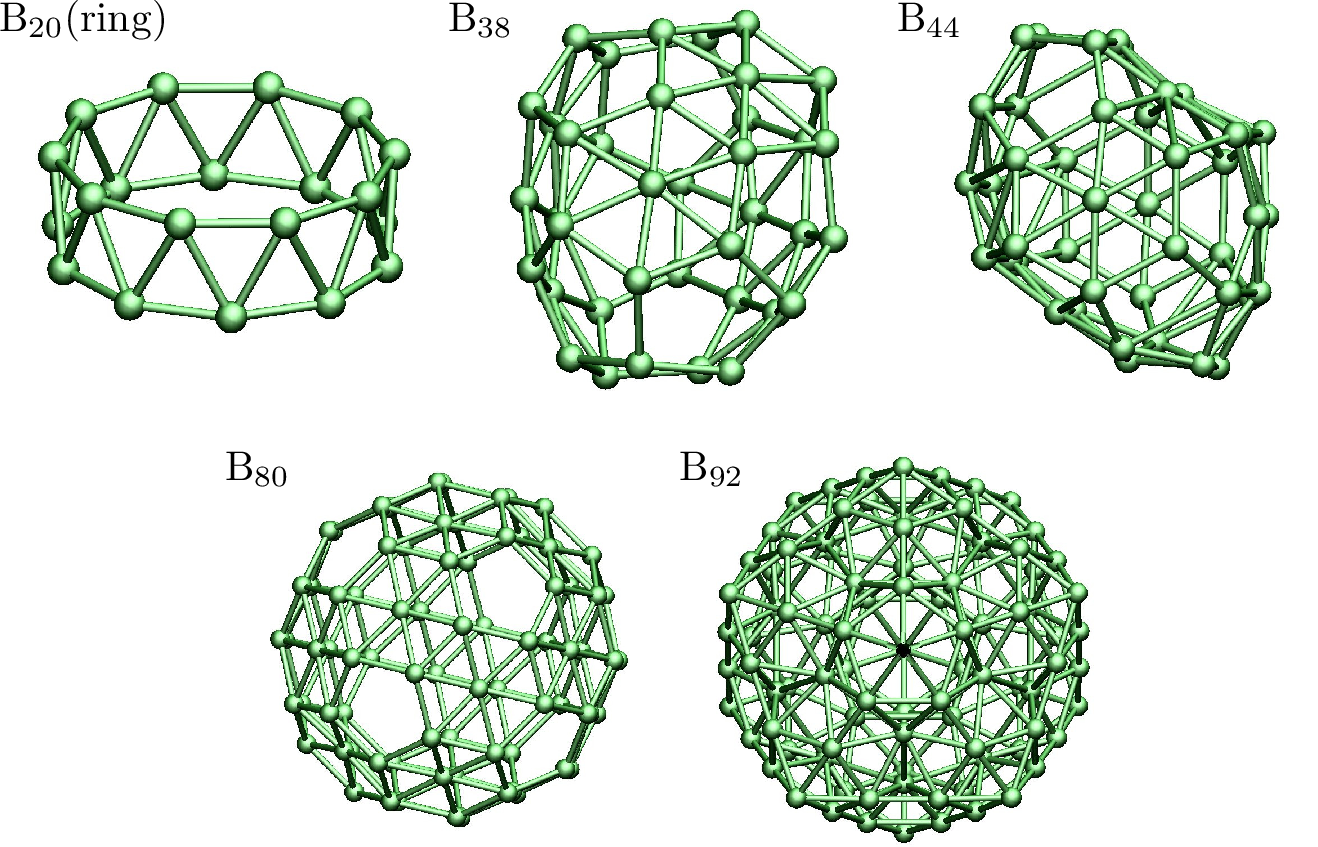
Real-space grids are a powerful alternative for the simulation of electronic systems. One of the main advantages of the approach is the flexibility and simplicity of working directly in real space where the different fields are discretized on a grid, combined with competitive numerical performance and great potential for parallelization. These properties constitute a great advantage at the time of implementing and testing new physical models. Based on our experience with the Octopus code, in this article we discuss how the real-space approach has allowed for the recent development of new ideas for the simulation of electronic systems. Among these applications are approaches to calculate response properties, modeling of photoemission, optimal control of quantum systems, simulation of plasmonic systems, and the exact solution of the Schrödinger equation for low-dimensionality systems.
Compressed Sensing for the Fast Computation of Matrices: Application to Molecular Vib...
Jacob Sanders
and 2 more
July 11, 2014
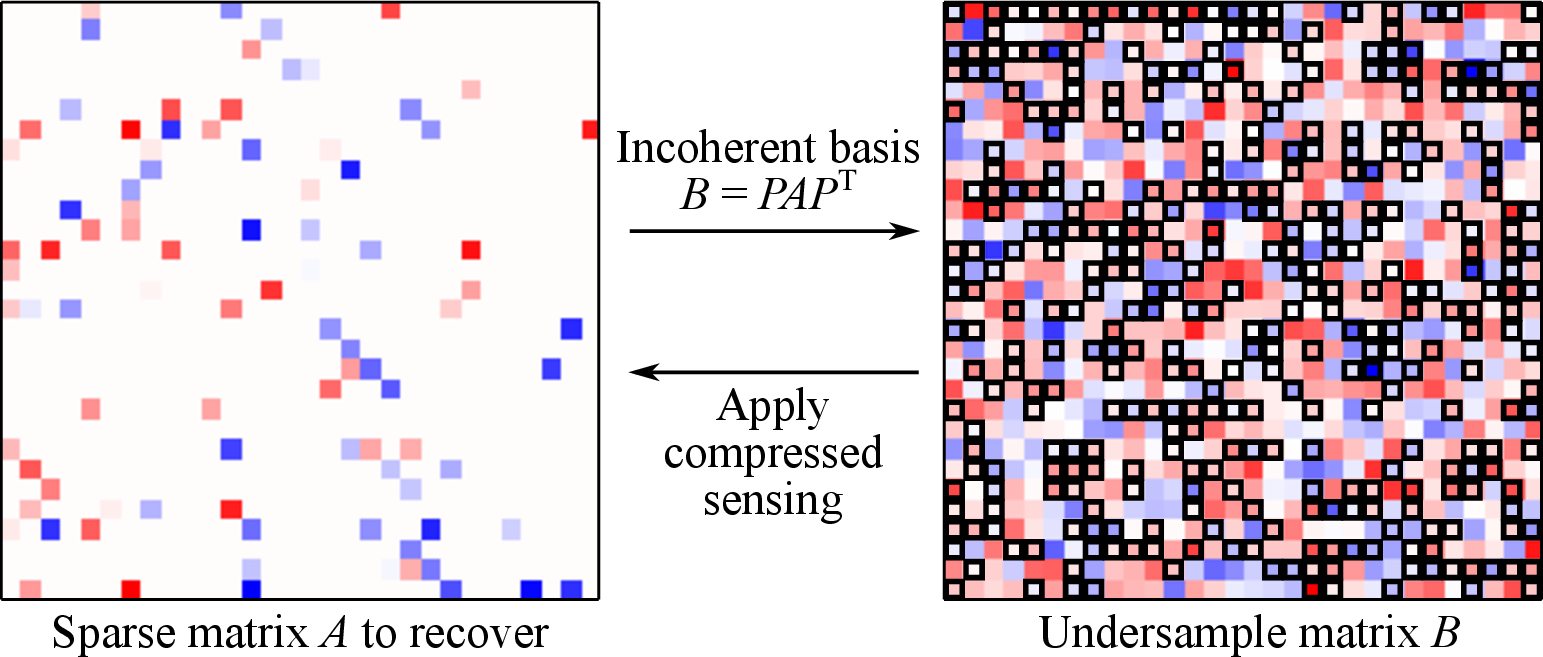
This article presents a new method to compute matrices from numerical simulations based on the ideas of sparse sampling and compressed sensing. The method is useful for problems where the determination of the entries of a matrix constitutes the computational bottleneck. We apply this new method to an important problem in computational chemistry: the determination of molecular vibrations from electronic structure calculations, where our results show that the overall scaling of the procedure can be improved in some cases. Moreover, our method provides a general framework for bootstrapping cheap low-accuracy calculations in order to reduce the required number of expensive high-accuracy calculations, resulting in a significant 3\(\times\) speed-up in actual calculations.