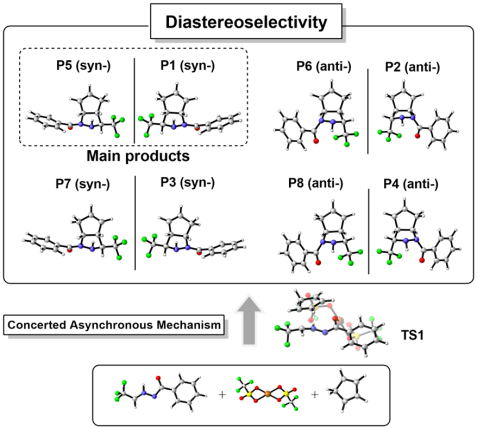
Pyrazolidines are very important compounds that widely exist in many natural products. Herein, we have employed high-level DFT calculations to systematically investigate the underlying mechanism of Cu(OTf)2 catalyzed [3+2] cycloaddition reactions that synthesis CF3substituted pyrazolidines. About eight possible initial configurations of the [3+2] reaction is considered and all relevant reactants, transition states and products are optimized. Based on these structures, IRC paths and the wavefunction analysis, we concluded that the Cu(OTf)2 catalyzed [3+2] cycloaddition follow a concerted asynchronous mechanism. The CN bond forms immediately after the formation of the CC bond. Among all eight reaction paths, the energy barrier for the [3+2] reaction that lead to the CF3substituted synpyrazolidine is the lowest one, ca. 3.2 kcal/mol, which might result in the diastereoselectivity that observed in experiment. We have also investigated the reaction processes that without Cu(OTf)2 molecule. The computational results indicate that the energy barriers that form the diastereoisomers are much closer and also larger than the Cu(OTf)2 catalyzed one. Therefore, Cu(OTf)2 catalyst plays an important role for the diastereoselectivity of the [3+2] cycloaddition reaction. Our present work not only gives the detail mechanism of the Cu(OTf)2 catalyzed [3+2] cycloaddition, but can also be helpful for the future designation of Cu(OTf)2 based cycloaddition processes.