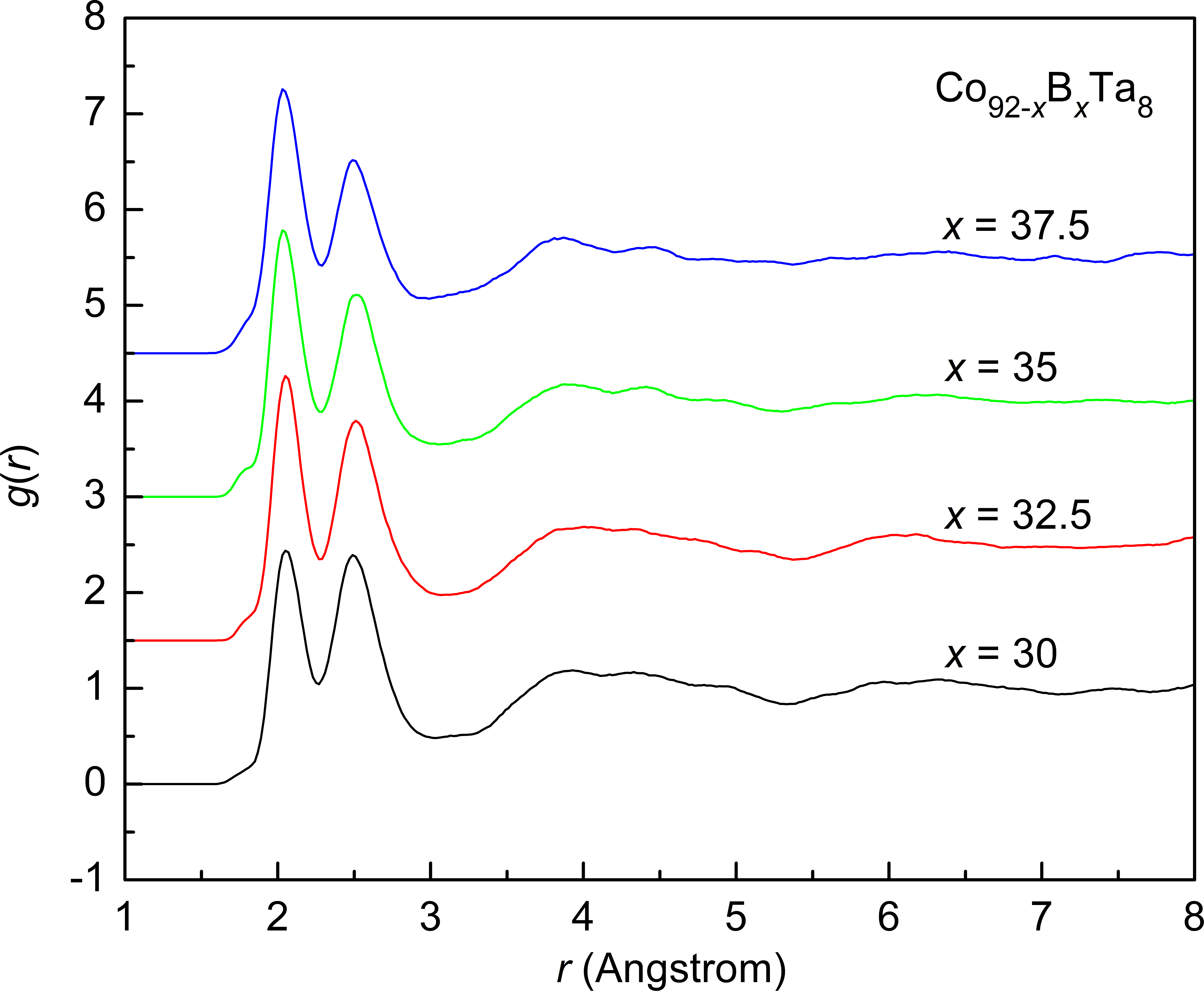
The ab initio molecular dynamics simulations are performed to study the atomic structures of Co92-xBxTa8 (x = 30, 32.5, 35, 37.5, at.%) glassy alloys. The result shows that the local packing of B-centered clusters is more efficient than that for Co- and Ta-centered clusters. It is also found that B-centered clusters are the primary structure-forming clusters. The Kasper polyhedra with a Voronoi index of <0 3 6 0> and <0 2 8 0> are dominant in B-centered clusters. Specially, the <0 3 6 0> clusters can form a robust network structure, which plays a key role in mechanical properties. Such a network structure has a higher activation barrier for structural rearrangement and a better resist to plastic flow. Thus, the increase in the fraction of <0 3 6 0> with B content would result in an increase in yield strength as well as a sharp decrease in compression plasticity.