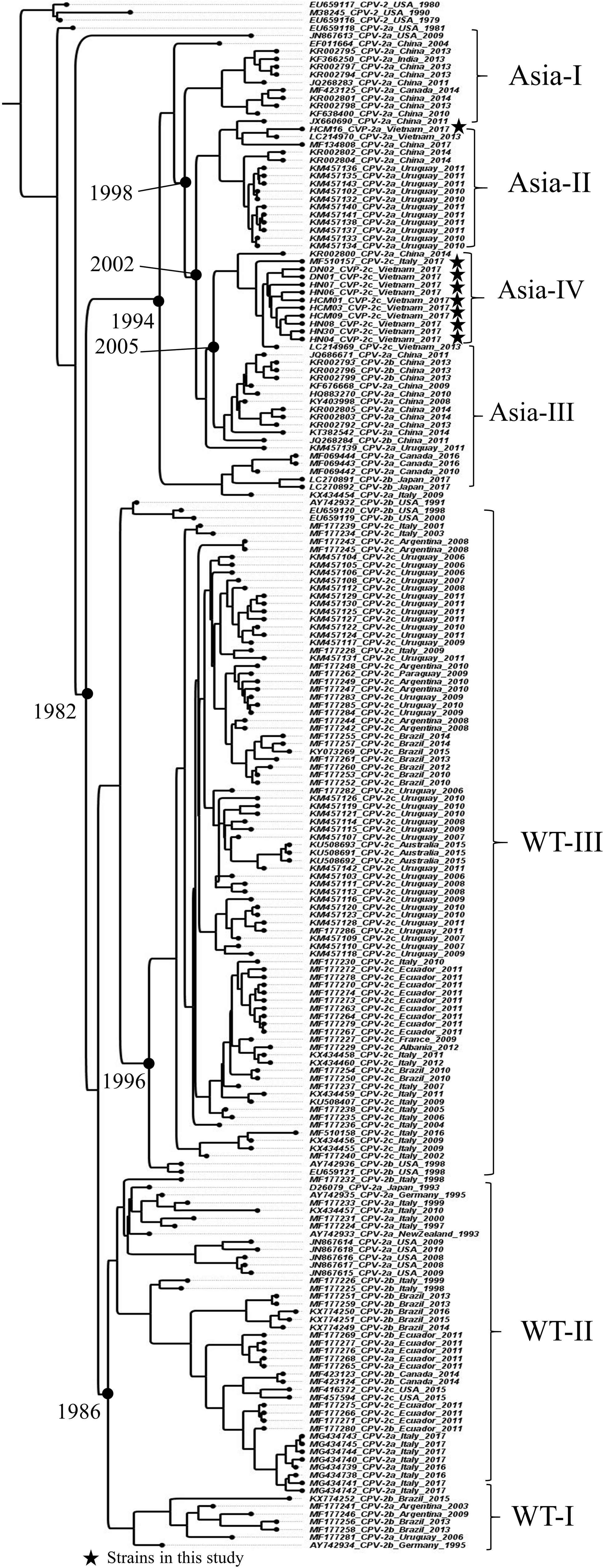
Canine parvovirus type 2 (CPV-2) is a small, single-stranded DNA virus causing fatal hemorrhagic enteritis in dogs. Currently, CPV-2 has been classified into CPV-2a, CPV-2b, and CPV-2c based on genetic variation in the VP2 gene. The CPV-2c variant has become ubiquitous worldwide and gained attention for monitoring parvoviral evolution. In this study, we characterized the full-length genome sequences of CPV-2c isolates obtained from 59 dogs in Vietnam. Molecular analysis revealed that Vietnamese CPV-2c shared a common evolutionary pattern with the Asian CPV-2 clade, which is marked by genetic signature patterns in the structural and nonstructural proteins. In addition, these Vietnamese CPV-2c strains exhibited unique Thr112Ile and Ile447Met mutations in the VP1 and VP2 sequence, respectively. Interestingly, phylogenetic analysis indicated that the mutations of amino acid residues in both the structural and nonstructural genes have contributed to the emergence of a new clade, designated here as the Asia-IV clade. The substitution rates, estimated from a dataset containing 199 sequences over the last 40 years, confirmed that CPV-2 showed a high rate of nucleotide substitution, at about 2.49 x 10-4 nucleotide substitutions per site per year (nt/s/y), with VP1/2 and NS1/2 estimates of 3.06 x 10-4 and 3.16 x 10-4 nt/s/y, respectively. Even though no evidence of genetic recombination in these Vietnamese CPV-2c strains was established, potential positive selection sites were observed in both the structural and nonstructural genes, suggesting the viral evolutionary process has occurred in both the structural and nonstructural proteins. Genetic and evolutionary analysis of the full-length genome sequence is necessary to gain evolutionary insight of CPV-2. Further studies are needed to elucidate the potential role of these observed mutations in the novel Asia-IV clade.