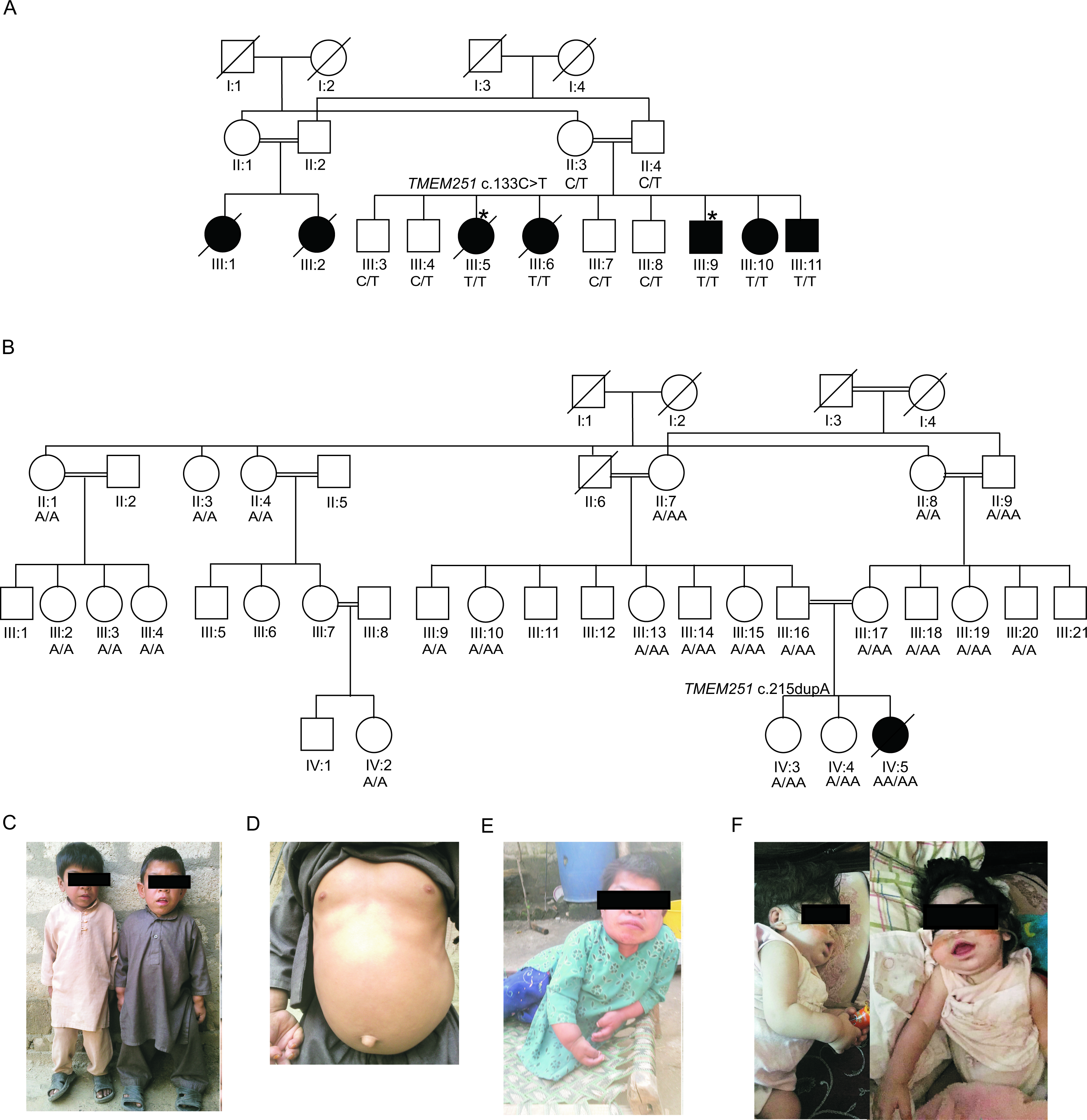
Skeletal dysplasias are a heterogeneous group of disorders ranging from mild to lethal skeletal defects. We investigated two unrelated families with individuals presenting with a severe skeletal disorder. In family NMD02, affected individuals had a dysostosis multiplex-like skeletal dysplasia and severe short stature (<-8.5 SD). They manifested increasing coarse facial features, protruding abdomens and progressive skeletal changes, reminiscent of mucopolysaccharidosis. The patients gradually lost mobility and the two oldest affected individuals died in their twenties. The affected child in family ID01 had coarse facial features and severe skeletal dysplasia with clinical features similar to mucopolysaccharidosis. She had short stature, craniosynostosis, kyphoscoliosis and hip-joint subluxation. She died at the age of 5 years. Whole-exome sequencing identified two homozygous variants c.133C>T; p.(Arg45Trp) and c.215dupA; p.(Tyr72Ter) respectively, in the two families, affecting an evolutionary conserved gene TMEM251. Immunofluorescence and confocal studies on Human Osteosarcoma cells indicated that TMEM251 localized to the Golgi complex and plasma membrane. However, p.Arg45Trp mutant TMEM251 protein was targeted less efficiently to the membranes and the localization was punctate. Tmem251 knockdown by siRNA induced dedifferentiation of rat primary chondrocytes. Our work implicates TMEM251 in the pathogenesis of a novel disorder and suggests its potential function in chondrocyte differentiation.