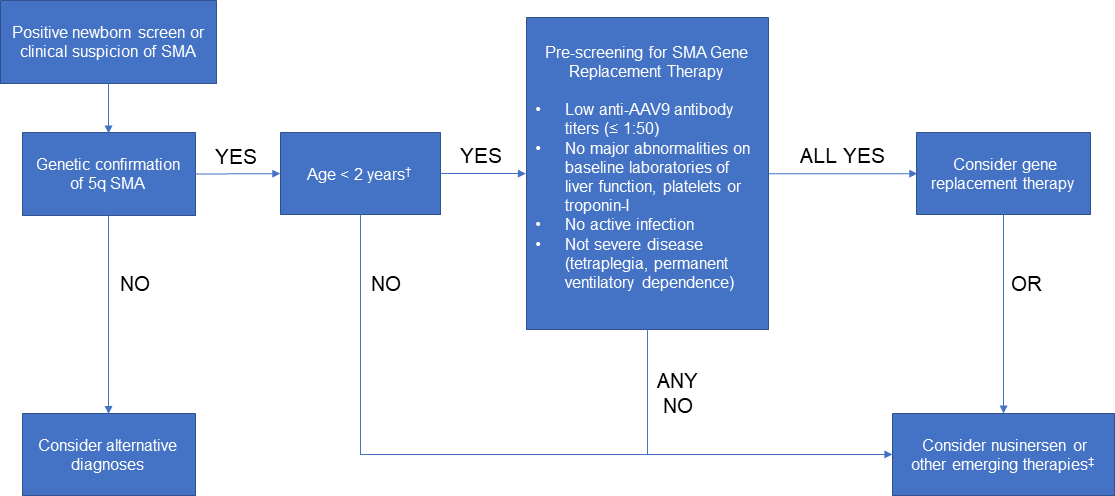
Both 5q-linked spinal muscular atrophy (SMA) and Duchenne muscular dystrophy (DMD) are fatal monogenic neuromuscular disorders caused by loss-of-function mutations. SMA is an autosomal recessive disorder affecting motor neurons that is typically caused by homozygous whole-gene deletions of SMN1. DMD is an X-linked recessive muscle disease most often due to exon deletions, but also duplications and smaller sized variants within the DMD gene. Gene replacement therapy offers the opportunity to correct the underlying genetic defect by the introduction of a functional gene. We review the transformative work from clinical trials to United States Food and Drug Administration approval of onasemnogene abeparvovec-xioi in SMA and its application in clinical practice and the early results of microdystrophin delivery in DMD. We also review the introduction of antisense oligonucleotides to alter pre-mRNA splicing to promote exon inclusion (as in nusinersen in SMA) or exclusion (as in eteplirsen in DMD) into neuromuscular therapeutics. There are multiple promising novel genetically mediated therapies on the horizon, which in aggregate point towards a hopeful future for individuals with SMA and DMD.