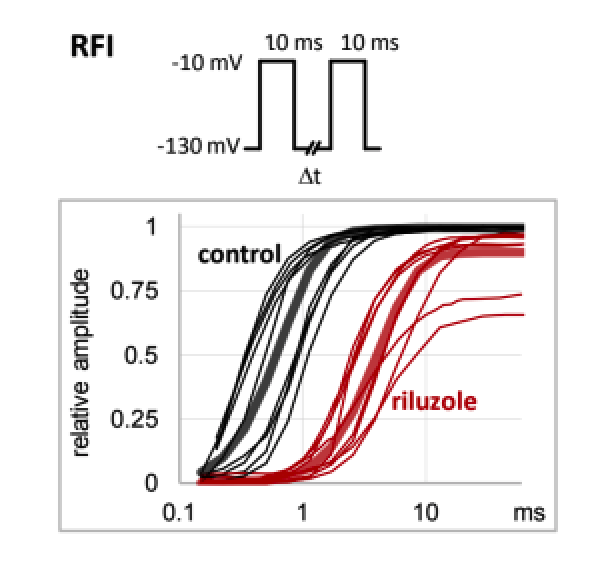
Hyperexcitability-related diseases include epilepsies, pain syndromes, neuromuscular disorders, and cardiac arrhythmias. Sodium channel inhibitors can be used to treat these conditions, however, their applicability is limited by their nonspecific effect on physiological function. They act by channel block (obstructing ion conduction, since the binding site is within the channel pore), and by modulation (delaying recovery from the non-conducting inactivated state). Channel block inhibits healthy and pathological tissue equally, while modulation can preferentially inhibit pathological activity. Therefore, an ideal sodium channel inhibitor drug would act by modulation alone. Unfortunately, thus far no such drug has been known to exist. Here we present evidence that riluzole acts by this “ideal” mechanism, “non-blocking modulation” (NBM). We propose that, being a relatively small molecule, riluzole is able to stay bound to the binding site, but nonetheless stay off the conduction pathway, by residing in one of the “fenestrations” (cavities connecting the central cavity to the membrane phase). Using precisely timed UV pulses to photolabel specific conformations of the channel, we show that association to the local anesthetic binding site requires prior inactivation. We discuss why kinetics of binding is crucial for selective inhibition of pathological activity, and how the NBM mechanism can be recognized using a special voltage- and drug application-protocol. Our results identify riluzole as the prototype of this new class of sodium channel inhibitors. Drugs of this class are expected to selectively prevent hyperexcitability, while having minimal effect on cells firing at a normal rate from a normal resting potential.