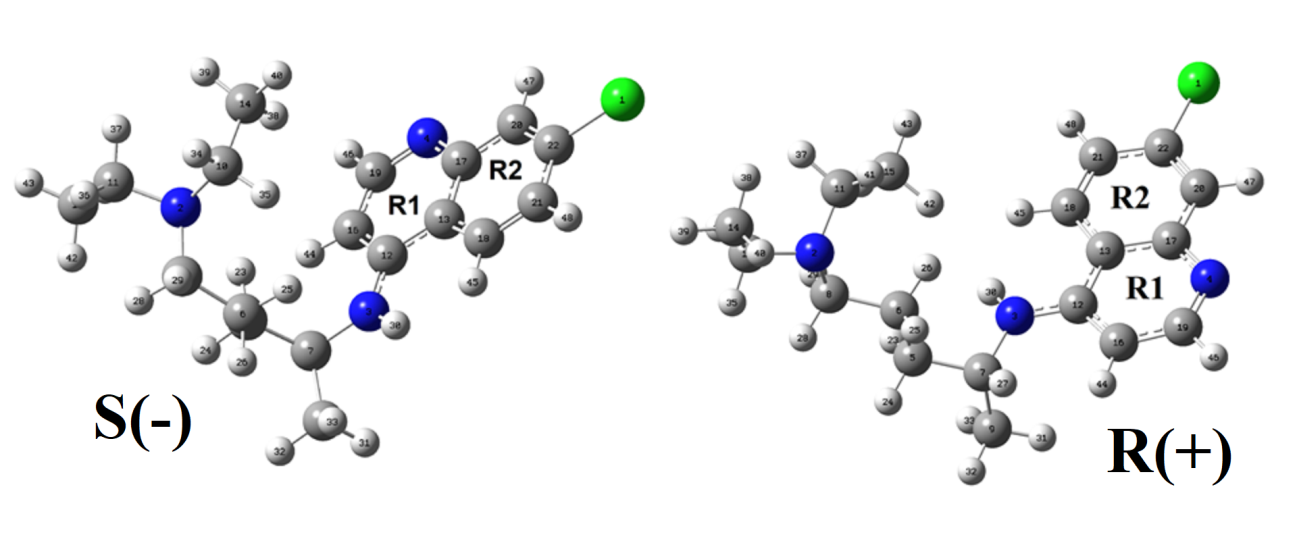
Structural, electronic, topological, vibrational and molecular docking studies have been performed for both enantiomeric S(-) and R(+) forms of potential antiviral to COVID-19 chloroquine (CQ) combining DFT calculations with SQMFF methodology. Hybrid B3LYP/6-311++G** calculations in gas phase and aqueous solution predict few energy differences between both forms. Solvation energies of S(-) and R(+) form are predicted in -55.07 and 59.91 kJ/mol, respectively. Low solvation energies of both forms are justified by the presence of only four donor and acceptor H bonds groups, as compared with other antiviral agents. MK charges on the Cl1, N2, N3 and N4 atoms and AIM calculations could support the high stability of R(+) form in solution according to the higher reactivity predicted for the S(-) form in this medium. Antiviral to COVID-19 niclosamide shows higher reactivity than both forms of CQ. Complete vibrational assignments of 153 vibration modes for both forms and scaled force constants have been reported here. Reasonable concordances were found between predicted and available 1H-NMR, 13C-NMR and UV-Vis spectra. Additionally, NMR and UV-visible spectra suggest the presence of two forms of CQ in solution. A molecular docking study was performed to identify the potency of inhibition of Chloroquine molecule against COVID-19 virus