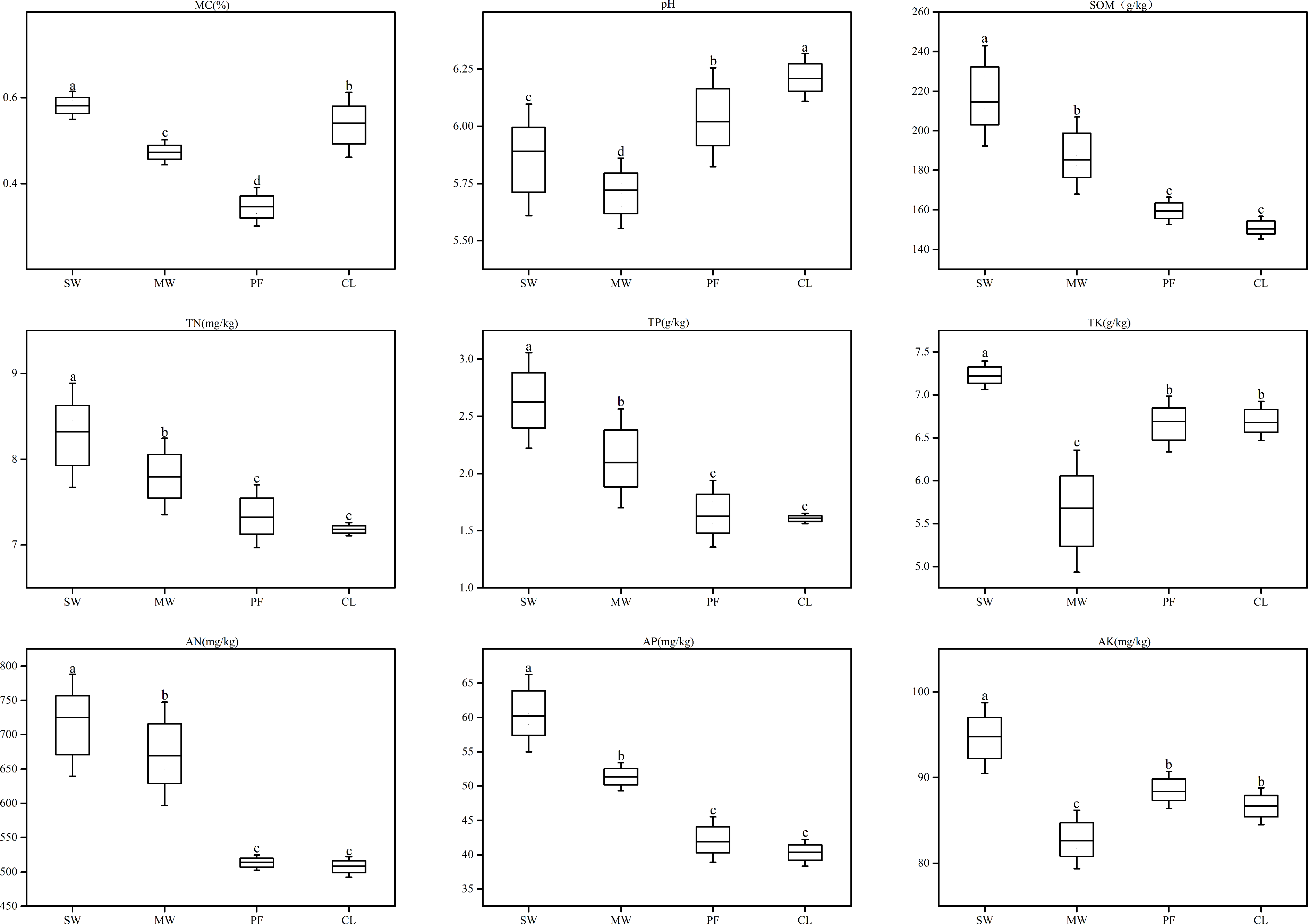
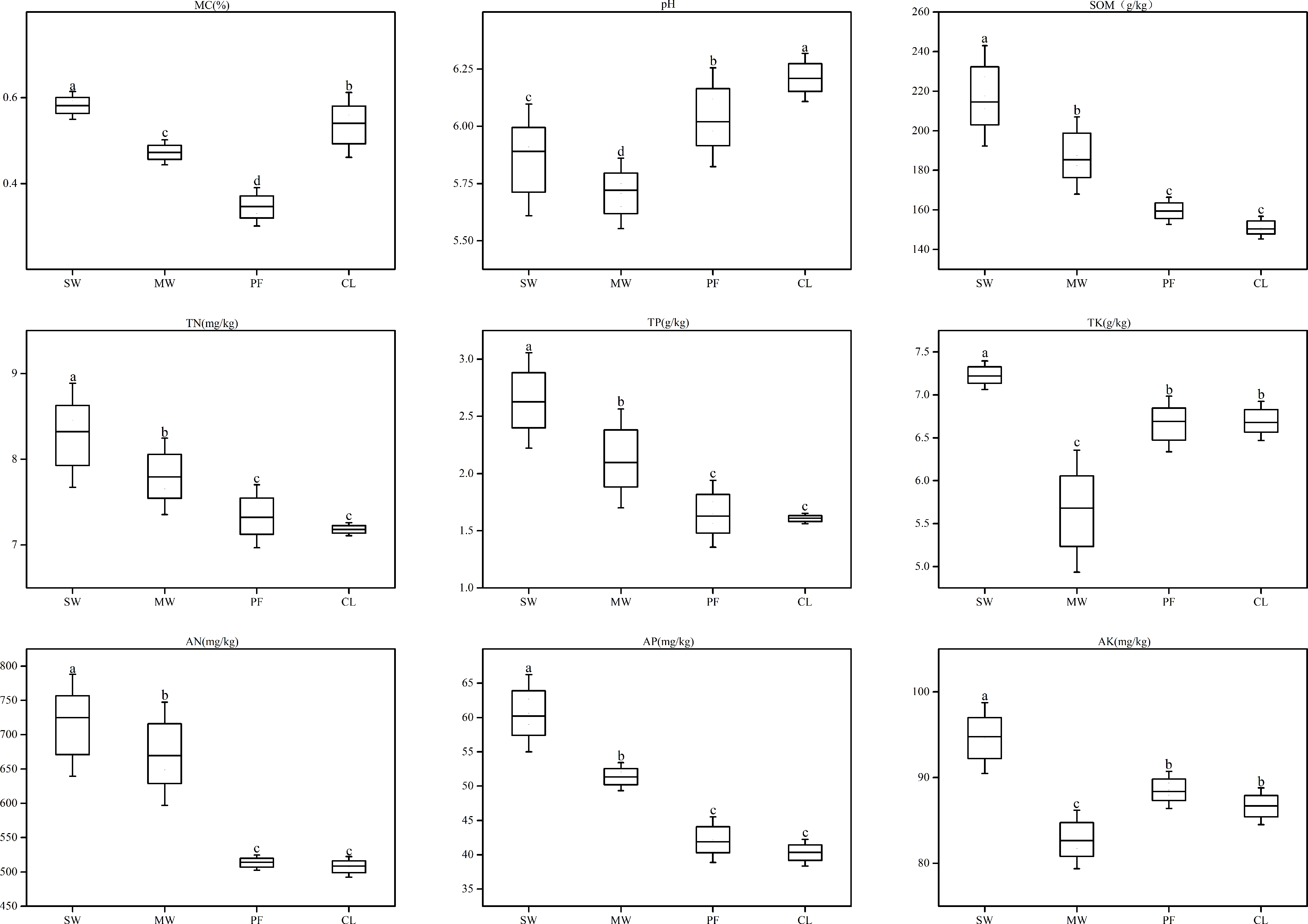
Sanjiang Plain is the largest area of freshwater wetland in China. Due to agricultural development, a large volume of groundwater in this area has been extracted over the last few decades, resulting in wetland degradation. In order to provide information for the development and protection of wetland ecosystem, investigations examining processes of wetland degradation are important. The aim of this work is to assess the impacts of wetland degradation on the communities of soil microbial community under four different types of degradation wetland including swamp meadow (SW), meadow wetland (MW), paddy farmland (PF), and cropland (CL) in Sanjiang Plain. Using both 16S and ITS rRNA gene amplicon sequencing to evaluate the fungal and bacterial diversity and composition. The dominant fungal phyla and bacterial were Ascomycota and Proteobacteria in this study, respectively. In addition, wetland degradation remarkably augmented the partial affluence of Chloroflexi and Gemmatimonadetes, but the partial affluence of Proteobacteria and Verrucomicrobia significantly diminished. Bacterial Shannon index of SW was lower than those in other sites. While, fungal diversity had no significant differences under different types of degradation wetland. Along with the wetland degradation, such differential reactions of the dominant phyla microbial and diversity were notably coordinated with TP, TK, AK, and SOM, which were the most essential criteria influencing the soil microbial communities. Generally, these outcomes suggested that wetland degradation could result in variations in soil microbial community composition structure. These changes could be used as an early warning signal for the degradation wetland in Sanjiang Plain.