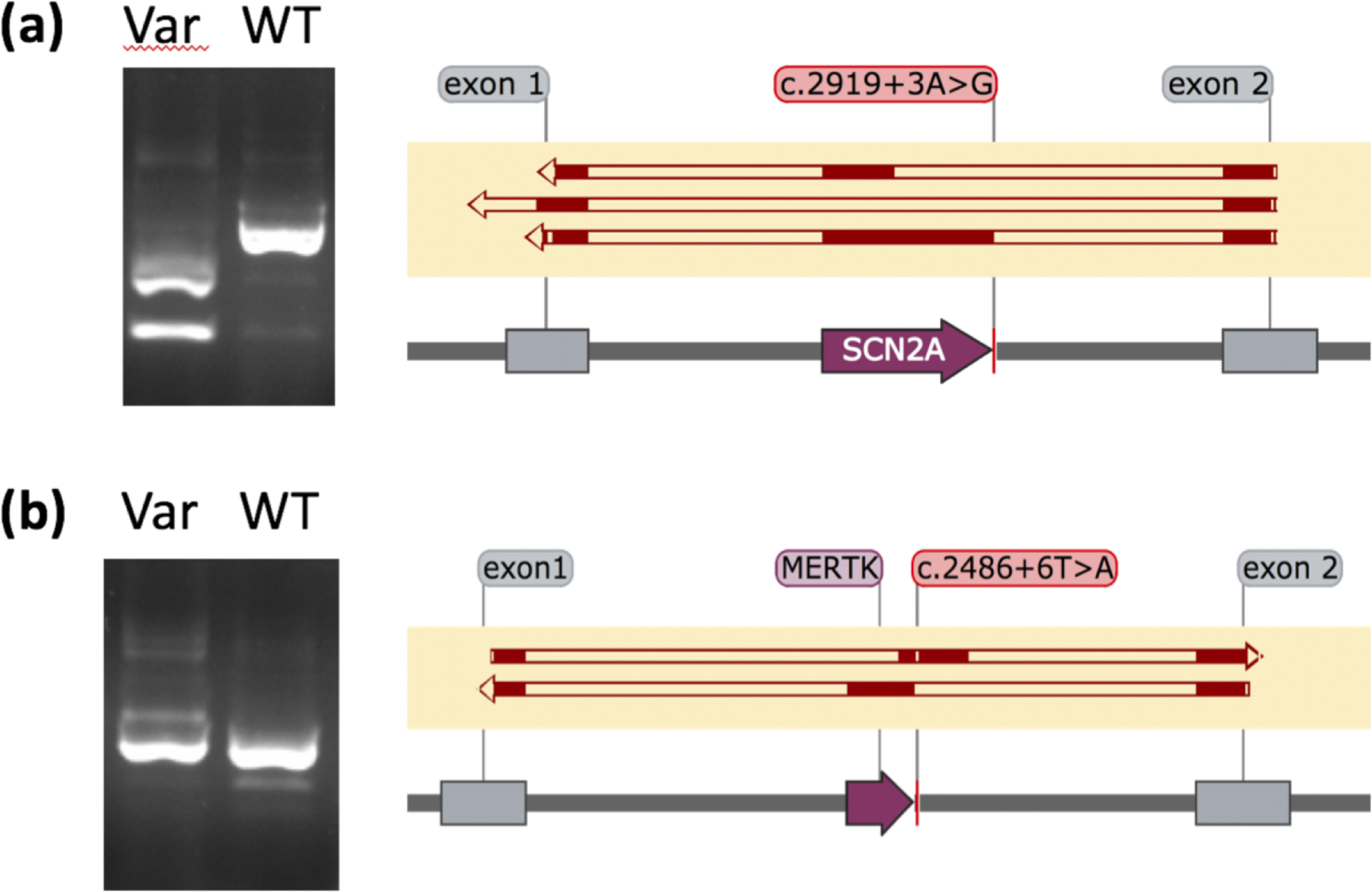
The development of computational methods to assess pathogenicity of pre-messenger RNA splicing variants is critical for diagnosis of human disease. We assessed the capability of eight algorithms, and a consensus approach, to prioritize 250 variants of uncertain significance (VUS) that underwent splicing functional analyses. It is the capability of algorithms to differentiate VUSs away from the immediate splice site as ‘pathogenic’ or ‘benign’ that is likely to have the most substantial impact on diagnostic testing. We show that SpliceAI is the best single strategy in this regard, but that combined usage of tools using a weighted approach can increase accuracy further. We incorporated prioritization strategies alongside diagnostic testing for rare disorders. We show that 15% of 2783 referred individuals carry rare variants expected to impact splicing that were not initially identified as ‘pathogenic’ or ‘likely pathogenic’; 1 in 5 of these cases could lead to new or refined diagnoses.