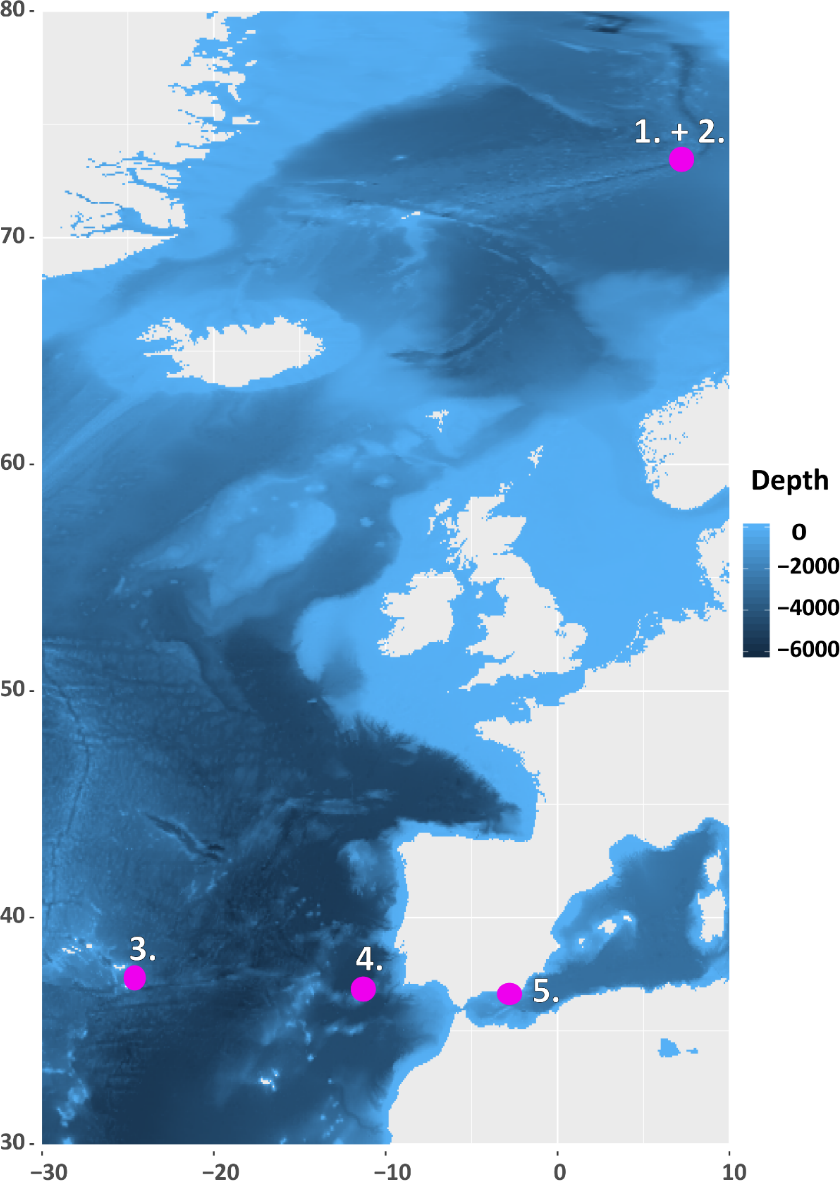
Biodiversity inventory remains limited in marine systems due to unbalanced access to the three ocean dimensions. The use of environmental DNA (eDNA) for metabarcoding allows fast and effective biodiversity inventory and is forecast as a future biodiversity research and biomonitoring tool. However, in poorly understood ecosystems, eDNA results remain difficult to interpret due to large gaps in reference databases and PCR bias limiting the detection of some major phyla. Here, we aimed to circumvent these limitations by avoiding PCR and recollecting larger DNA fragments to improve assignment of detected taxa through phylogenetic reconstruction. We applied capture by hybridization (CBH) to enrich DNA from deep-sea sediment samples and compared the results with those obtained through an up-to-date metabarcoding PCR-based approach (MTB). Originally developed for bacterial communities by targeting 16S rDNA, the CBH approach was applied to 18S rDNA to improve the detection of species forming benthic communities of eukaryotes, with particular focus on metazoans. The results confirmed the possibility of extending CBH to metazoans with two major advantages: i) CBH revealed a broader spectrum of prokaryotic, eukaryotic, and particularly metazoan diversity, and ii) CBH allowed much more robust phylogenetic reconstructions of full-length barcodes with up to 1900 base pairs. This is particularly important for taxa whose assignment is hampered by gaps in reference databases. This study provides a database and probes to apply 18S CBH to diverse marine systems, confirming this promising new tool to improve biodiversity assessments in data-poor ecosystems like those in the deep sea.