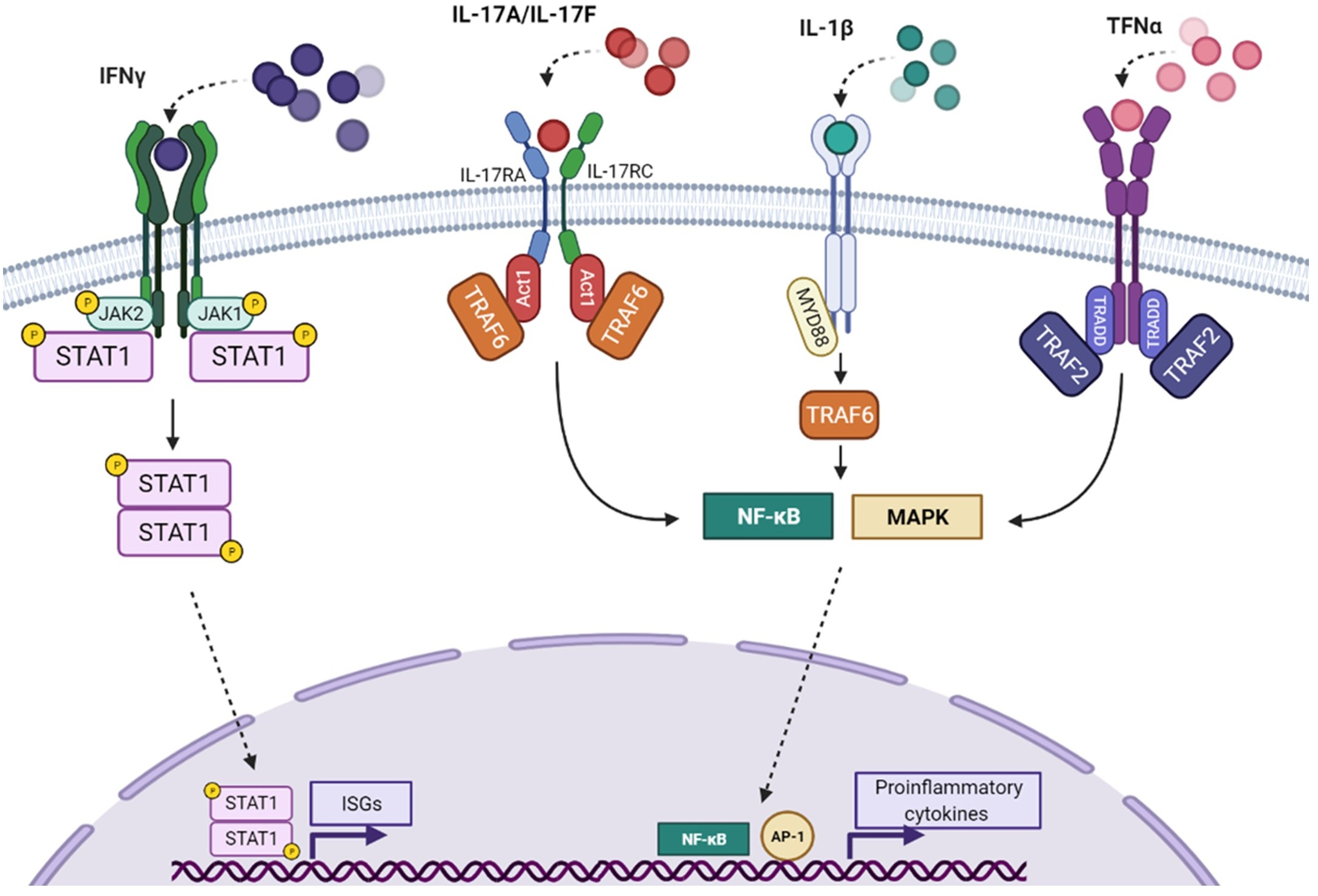
Background: Inherited chronic mucocutaneous candidiasis (CMC) is often caused by inborn errors of immunity, impairing the response to, or the production of IL-17A and IL-17F. About half of the cases carry STAT1 gain-of-function (GOF) mutations. Only few patients have been reported with mutations of TRAF3IP2, a gene encoding the adaptor ACT1 essential for IL-17-receptor(R) -signaling. We investigated a 10-year-old girl with CMC, carrying a heterozygous variant of STAT1 and compound heterozygous variants of TRAF3IP2. Methods: By flow cytometry STAT1 levels and phosphorylation (CD14+) as well as IL-17A-, IL-22-, IFN-γ- and IL-4-production (memory CD4+ T cells) were determined. ACT1 expression and binding to IL-17RA by western blot and co-immunoprecipitation in HEK-293T cells transfected with plasmids encoding wild-type or mutant HA-tagged ACT1 and Flag-IL-17RA. We evaluated IL-17A response using an NF-κB-driven luciferase reporter system in HEK-293T cells, and by measuring GRO-α secretion by fibroblasts. Results: A likely non-pathogenic STAT1 variant (c.1363G>A/p.V455I) was identified by next generation sequencing., STAT1 expression and phosphorylation upon IFN- were normal. We also found compound heterozygous variants (c.1325A>G/p.D451G and c.1335delA/p.K454fs11*) of TRAF3IP2. By overexpression, despite normal protein expression, and impaired (K454fs11*) or normal (D451G) interaction with IL-17RA, both mutant alleles resulted in an impaired NF-κB-activation. Patient’s fibroblasts displayed abolished GRO-α secretion upon IL-17A. Finally, ex vivo CD4+ T cells showed increased IL-17A, IL-22, and IL-4, and normal-low IFN-γ expression upon stimulation. Conclusion: We identify novel compound heterozygous variants of TRAFP3IP2 causing autosomal recessive ACT1 deficiency in a child with CMC, and provide a review of the current literature.