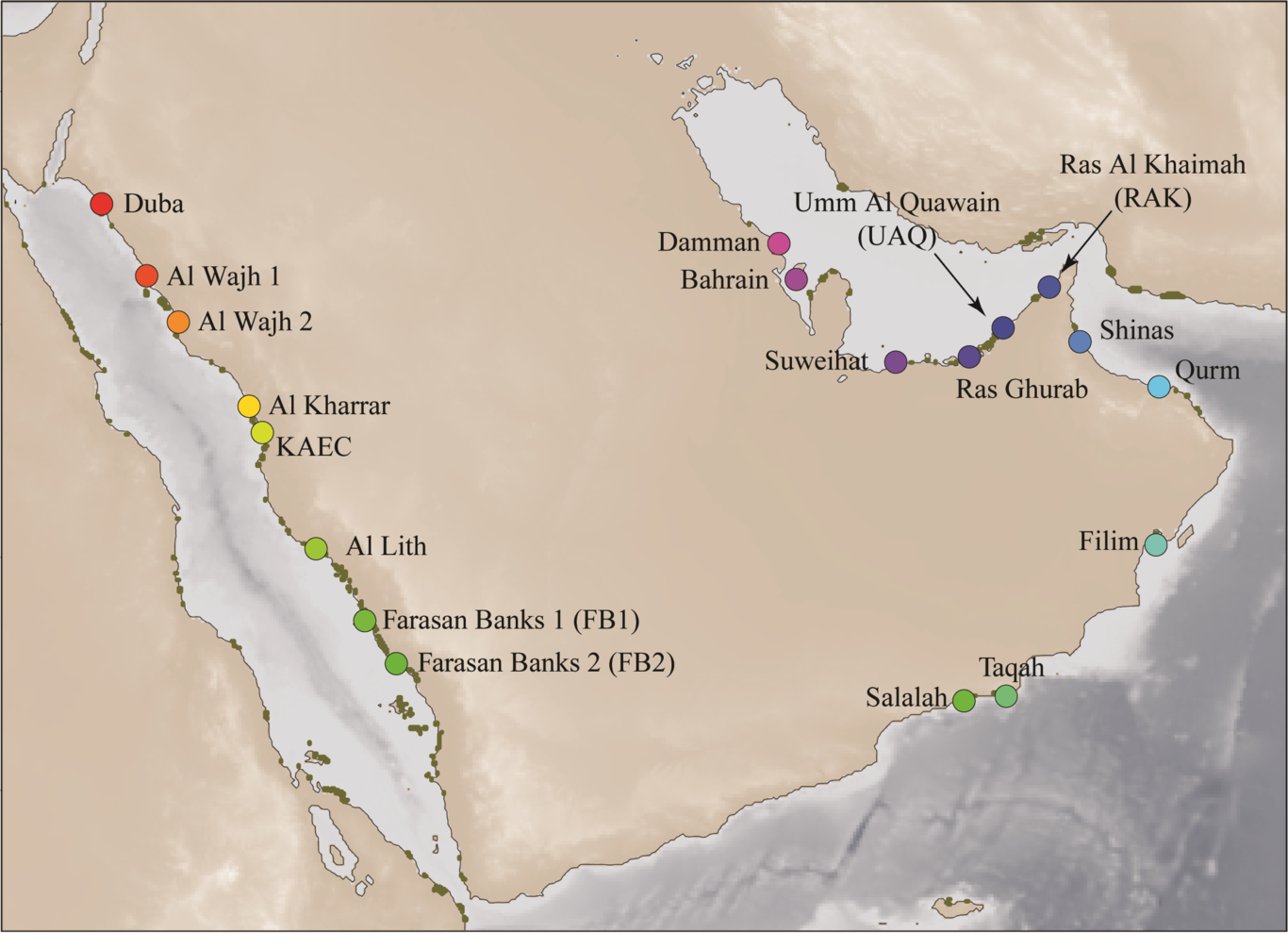
Running title: Evolution of Arabian mangrovesGuillermo Friis*, Edward G. Smith, Catherine E. Lovelock, Alejandra Ortega, Alyssa Marshell, Carlos M. Duarte, John A. Burt*Corresponding author: Center for Genomics and Systems Biology, New York University — Abu Dhabi, PO Box 129188, Abu Dhabi, United Arab Emirates; Email: [email protected]; Tel: +97126286739. AbstractBiological systems occurring in ecologically heterogeneous and spatially discontinuous habitats provide an ideal opportunity to investigate the relative roles of neutral and selective factors in driving lineage diversification. The gray mangroves (Avicennia marina) of Arabia occur at the northern edge of the species’ range and are subject to variable, often extreme, environmental conditions, as well as historic large fluctuations in habitat availability and connectivity resulting from Quaternary glacial cycles. Here, we analyze fully sequenced genomes sampled from 19 locations across the Red Sea, the Arabian Sea and the Persian/Arabian Gulf (PAG) to reconstruct the evolutionary history of the species in the region, and to identify adaptive mechanisms of lineage diversification. Population structure and phylogenetic analyses revealed marked genetic structure correlating with geographic distance and highly supported clades among and within the seas surrounding the Arabian Peninsula. Demographic modelling showed times of divergence consistent with recent periods of geographic isolation and low marine connectivity during glaciations, suggesting the presence of (cryptic) glacial refugia in the Red Sea and the PAG. Significant migration was detected within the Red Sea and the PAG, and across the Strait of Hormuz to the Arabian Sea, suggesting gene flow upon secondary contact among populations. Genetic‐environment association analyses revealed high levels of adaptive divergence, and detected signs of multi-loci local adaptation driven by temperature extremes and hypersalinity. These results support a process of rapid diversification resulting from the combined effects of historical factors and ecological selection, and reveal mangrove peripheral environments as relevant drivers of lineage diversity.IntroductionLineage diversification involves both neutral and selective factors, and elucidating their relative roles in the process of evolutionary divergence is essential to understand the mechanisms underlying the early stages of speciation (Coyne & Orr, 2004; Nosil, 2012). Evolutionary divergence may result from the accumulation of genetic differences caused by drift in geographic isolation or isolation-by-distance (IBD, Wright, 1943, 1946), a mode of divergence driven by neutral factors (Mayr, 1954, 1963). In turn, geographic variation in environmental conditions can result in divergent selection, the diversifying process that drives ecological speciation (Coyne & Orr, 2004; Darwin, 1859; Nosil, 2012). In ecological speciation models, reproductive barriers arise as a by-product of cumulative, ecologically adaptive changes (Mayr, 1947; Rundle & Nosil, 2005; Schluter, 2000), enabling genome‐wide differentiation at both neutral and selected loci (Funk, Egan, & Nosil, 2011; Nosil, Egan, & Funk, 2008; Shafer & Wolf, 2013; Wang & Bradburd, 2014). Ecological speciation in geographic isolation is theoretically uncontroversial, and deemed common in nature as a mechanism maintaining lineage diversity upon secondary contact (Keller & Seehausen, 2012; Nosil, 2012; Rundle & Nosil, 2005). However, whether environment-driven processes of lineage diversification occur frequently in nature in the absence of long-term geographic isolation and reduced gene flow remains debated in evolutionary research (Bolnick & Fitzpatrick, 2007; Fitzpatrick, Fordyce, & Gavrilets, 2008; Foote, 2018). The interactions between selection and the stochastic effects derived from processes such as founder events, bottlenecks and genetic drift also remain unclear, and difficult to assess in natural systems (Barton & Charlesworth, 1984; Burri et al., 2015; Kliber & Eckert, 2005). Biological systems occurring at the species' range edges, which are frequently extreme and environmentally diverse habitats, are suitable models to investigate questions related to lineage diversification. The environment at the edges of species’ range tends to be stressful and spatially discontinuous, as well as temporally unstable (Lesica & Allendorf, 1995), often resulting in dynamic settings of multiple isolated populations subject to strong differential selection. The severe and stochastic character of peripheral environments is hypothesized to generate strong selective interplay between adaptation and neutral processes (Hardie & Hutchings, 2010), providing an ideal opportunity for speciation research. One such system is provided by gray mangrove populations in the Arabian Peninsula (Avicennia marina var. marina). The gray mangrove has the broadest distribution of any mangrove species (Hogarth, 2015; Spalding, Kainuma, & Collins, 2010; Tomlinson, 2016), extending across the Indian Ocean and into the West Pacific as far as Japan and New Zealand (Fouda & AI-Muharrami, 1996; Khalil, 2015; C. Sheppard et al., 2010; Spalding et al., 2010). They present several morphological and physiological adaptations to their harsh intertidal habitat (Tomlinson, 2016), which makes them a compelling model for the study of functional genes and biological pathways involved in selection and stress tolerance (Urashi, Teshima, Minobe, Koizumi, & Inomata, 2013; Xu et al., 2017). The Arabian Peninsula represents one of the northernmost edges of the species’ distribution (NC Duke, 1991; Spalding et al., 2010; Tomlinson, 2016), as well as a stressful habitat characterized by extreme temperatures, aridity, and often extreme salinity, factors known to be limiting for mangrove growth (Ball, 1988; Lovelock, Krauss, Osland, Reef, & Ball, 2016; Charles Sheppard, Price, & Roberts, 1992). Arabian marine domains are also environmentally diverse both within, and between, the main water bodies bordering the peninsula, which define three main biogeographic regions: (i) the Red Sea, where the marine system presents opposing gradients of salinity and temperature, with the highest temperature and lowest salinity in shallow southern basin, while the north has cooler temperatures but high salinity as a result of limited precipitation and high evaporation (Anton et al., 2020; Carvalho, Kürten, Krokos, Hoteit, & Ellis, 2019); (ii) the Persian/Arabian Gulf (referred to as ‘PAG’ hereafter) to the northeast of the Arabian Peninsula, where populations are subject to arid (<250 mm/year) to hyper-arid (<100 mm/year) rainfall regimes, and experience the widest range of air temperatures in the region throughout the year (Böer, 1997; Whitford & Duval, 2019); and (iii) the Arabian Sea and Sea of Oman, which in contrast with former biogeographic regions, has normal oceanic salinity, and summer temperatures that are buffered by cold-water upwelling as a result of the Indian Ocean monsoon, resulting in more moderate environmental conditions (Claereboudt, 2019). The Arabian Peninsula has experienced large fluctuations in spatial and environmental conditions throughout glacio-eustatic cycles that largely impacted the biodiversity of the region, in particular the enclosed water bodies of the Red Sea and the PAG (DiBattista, Choat, et al., 2016). Throughout the last 400,000 years the Red Sea has remained connected to the Indian Ocean, yet cross-sectional area along the Strait of Bab al Mandab that connects these water bodies was, at times of glacial maxima, as low as 2% of that today, resulting in major increases in salinity and temperature within the Red Sea as well as near-complete isolation at times (Lambeck et al., 2011). For several sustained periods during the last two glacial cycles, the minimum channel width connecting the Red Sea to the Arabian Sea was less than 4 km wide and remained narrow whenever the local sea levels were 50 meters below current levels (Lambeck et al., 2011). In contrast, models show that the PAG was nearly completely drained during the peak of the last glaciation until c.a. 14,000 years ago (Lambeck, 1996). A marine incursion into the southern PAG basin started approximately 12,500 years ago, extending towards the northern basin over the following millennia, with the present day PAG shorelines forming just 6,000 years ago (Lambeck, 1996). In contrast, as an open ocean habitat, the Arabian Sea coast has only experienced vertical migration of sea levels during these glacial periods, without geographic isolation. The combination of extreme environmental conditions, differential changes in habitat and dynamic barriers to gene flow, makes the seas bordering the Arabian Peninsula one of the most variable marine environments in the world, with a high potential for speciation driven by both neutral and selective factors (DiBattista, Roberts, et al., 2016). Although the phylogenetic relationships for the varieties of A. marina and congeneric species have been reported for other regions (N. C. Duke, Benzie, Goodall, & Ballment, 1998; X. Li et al., 2016; Nettel, Dodd, Afzal‐Rafii, & Tovilla‐Hernández, 2008), the extensive gray mangrove populations from the Arabian coasts have rarely been included in reported DNA sequence-based analyses (see Al-Qthanin & Alharbi, 2020; N. C. Duke et al., 1998; X. Li et al., 2016; Maguire, Saenger, Baverstock, & Henry, 2000), so that their evolutionary origin and relationships remain largely unexplored. The specific drivers and molecular basis of local adaptation and lineage diversification in A. marina also remain understudied both in Arabia and across its entire distribution. Here, we used the Arabian gray mangrove complex to examine how extreme habitat conditions and heterogeneous spatial settings have shaped genetic diversity at the highly variable edge of the species’ range using whole genome and georeferenced environmental data. First, we analyzed patterns of population structure and reconstructed the evolutionary and demographic history of the species in the Arabian Peninsula. Two general competing hypotheses about the evolutionary history of the Arabian mangroves were tested in this study:(i) mangroves from the Red Sea and PAG were extirpated during the glacial cycles of the Pleistocene, followed by a recolonization after the last glacial maximum (LGM); and (ii) mangroves remained within the enclosed seas in glacial refugia during glacial periods, and expanded once sea levels rose. Second, we studied patterns of adaptive variability applying genotype-environment association (GEA) analysis. We used redundancy analysis combining environmental and single nucleotide polymorphisms (SNP) data to survey the genome and jointly identify environmental variables and functional genes potentially involved in local adaptation and lineage divergence. Materials and MethodsPopulation sampling We sampled a total of 200 individuals of Avicennia marina from 19 sites of the Arabian Peninsula coasts (var. marina, N = 190), and one site from Australia (var. australasica, N = 10) to be used as outgroup (Fig. 1A; Table S1, Supplementary Information). Leaf tissue was collected from trees separated by at least 20 meters and preserved in silica beads for up to ten days before extraction. Geographic coordinates for each one of the trees were recorded. Genomic DNA was extracted from ground leaf tissue using the DNeasy 96 plant kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Genome resequencing and variant callingIllumina paired-end 150 bp libraries with insert size equal to 350 bp were prepared and sequenced in a Novaseq platform. A total of 8 billion reads were produced resulting in a mean coverage per site and sample of 28X before filtering. Read quality was evaluated using FASTQC (Andrews, 2010) after sorting reads by individual with AXE version 0.3.3 (Murray & Borevitz, 2017). Trimming and quality filtering treatment was conducted using Trim Galore version 0.6.6 (Krueger, 2015) with parameters --stringency 1 --clip_R1 12 --clip_R2 12 --length 90, resulting in a set of reads ranging between 90 and 138 bp long. Reads were then mapped against the previously published reference genome for A. marina (Friis et al., 2020) using the mem algorithm in the Burrows-Wheeler Aligner (BWA; H. Li & Durbin, 2009) version 0.7.1.7. Read groups were assigned and BAM files generated with Picard Tools version 1.126 (http://broadinstitute.github.io/picard). Duplicates were marked also with Picard Tools v1.126. We used the HaplotypeCaller + GenotypeGVCFs tools from the Genome Analysis Toolkit (GATK; McKenna et al., 2010) version 4.1.8.1 to produce a set of SNPs in the variant call format (vcf). Using vcftools version 0.1.16 (Danecek et al., 2011), we retained biallelic SNPs excluding those out of a range of coverage between 4 and 50, or with a genotyping phred quality score below 40. Twenty-two samples presenting more than a 25% of missing data were discarded at this point. We then applied GATK generic hard-filtering recommendations consisting of QualByDepth (QD) > 2.0; FisherStrand (FS) < 60.0; RMSMappingQuality (MQ) > 40; MappingQualityRankSumTest (MQRankSum) > -12.5; ReadPosRankSum (RPRK) < -8.0; and StrandOddsRatio (SOR) > 3.0 (GATK Best Practices; Auwera et al., 2013; DePristo et al., 2011). The resulting dataset (hereafter referred to as ‘Full Dataset’) consisted of 178 individuals and 15,702,886 SNPs (Table 1; Table S2, Supporting Information) with a per-individual average coverage of 16.8 and a missing data rate of 0.11. The program KING version 2.2.7 (Manichaikul et al., 2010) was used to confirm the absence of close relatives in our set of sampled individuals. Widespread self-fertilization was also ruled out using the program RMES and 300 variant positions randomly selected (Table S3, Supporting Information). The ‘Full Dataset’ was further filtered and customized for downstream analyses (Table S2, Supporting Information) Population structure analysesTo explore genome-wide population structure in Arabian mangroves, we conducted a principal components analysis (PCA). After excluding the samples from Australia, SNP loci under linkage disequilibrium were filtered out from the ‘Full Dataset’ with bcftools version 1.12 (Danecek & McCarthy, 2017) applying and r2 limit of 0.2 in windows of 10K bp. Using vcftools, a threshold for SNPs showing highly significant deviations from Hardy-Weinberg equilibrium (HWE) with a p-value of 10-4 was also implemented to filter out false variants arisen by the alignment of paralogous loci. Positions with less than 75% of individuals genotyped for each population were also removed from the data matrix, along with those presenting a minor allele frequency (MAF) below 0.02. To fulfil neutrality assumptions in population structure analyses, we used PCADAPT version 5.2 (Luu, Bazin, & Blum, 2017) to detect and exclude sites putatively under selection, applying a q-value threshold of 0.05 , resulting in a final data matrix of 143,900 SNPs and 170 samples. The PCA was conducted with the R package SNPRelate version 1.26.0 (Zheng, 2012). Using the same dataset as in the PCA, we examined patterns of population divergence in Arabian mangroves using a sparse non-negative matrix factorization method (SNMF) as implemented in R package LEA version 2.0.0 (Frichot, Mathieu, Trouillon, Bouchard, & François, 2014). We selected SNMF for inferring genetic structure due to its efficiency and shorter computational time. We ran the program five times per K value, with K ranging from 2 to 20. Similarity scores among runs and graphics were computed with CLUMPAK version 1.1.2 (Kopelman et al. 2015). To further explore patterns of geographic variation in Arabian mangroves, the dataset used in the PCA and the SNMF analysis was also used to test for isolation-by-distance. We computed pairwise Nei’s genetic distance values with the StAMPP R package version 1.6.0 (Pembleton, Cogan, & Forster, 2013). By-sea, pairwise geographic distances were measured based on GIS data. A Mantel test was implemented using the vegan R package version 2.5-7 (Oksanen, Blanchet, Kindt, Legendre, & O’Hara, 2016), and significance was computed through 9,999 matrix permutations. A linear regression between pairwise Nei’s genetic distances and geographic distances was also implemented and plotted for visual inspection. In addition, we used StAMPP to compute pairwise FST (Weir & Cockerham, 1984) among sampled populations. Significance was tested by conducting 100 permutations.A hierarchical analysis of molecular variance (AMOVA) as described in Excoffier et al. (1992) was used to assess the genetic structure of Arabian mangroves using the pegas R package version 1.2 (Paradis, 2010). We grouped individuals into two levels: major clades identified in phylogenetic analyses (see results below), and sampled populations within clades. We also computed the observed (HO) and expected heterozygosity (HE), and the nucleotide diversity (π) for each sampling site using the population program from Stacks version 2.63 (Catchen, Hohenlohe, Bassham, Amores, & Cresko, 2013). For this analysis, we did not exclude the Brisbane population belonging to the Australian variety of the gray mangrove (A. marina var. australasica), yet applied the same filters than for the analyses above (SNP matrix = 113,706). Phylogenetic analysisA maximum likelihood phylogeny was produced using the program IQ-TREE version 2.1.3 (Nguyen, Schmidt, Von Haeseler, & Minh, 2015) based on the SNP dataset used for heterozygosity and diversity analysis. In this case, ambiguously constant sites (positions lacking homozygous representants of at least one of the two alleles) were excluded (SNP matrix = 29,433) to enable ascertainment bias correction. The generalized time-reversible (GTR) model was implemented. The Brisbane population was used as outgroup. Branch support was estimated using the ultrafast bootstrap approximation by Hoang, Chernomor, and Von Haeseler (2018) with 1,000 iterations. Phylogenetic relationships were also inferred using the SVDQuartets model (Chifman & Kubatko, 2014) as implemented in PAUP version 4a169. We evaluated all the possible quartets and assessed branch support using 1,000 bootstrap replicates. The Brisbane population was used to root the tree. Population and demographic history analyses We performed model comparisons under the likelihood framework developed in fastSIMCOAL2 version 2.7 (Laurent Excoffier, Dupanloup, Huerta-Sánchez, Sousa, & Foll, 2013) to estimate demographic parameters and date cladogenetic events among mangroves populations, and to test the competing hypotheses of colonization after the LGM versus potential isolation in glacial refugia in the enclosed seas around Arabia. Three sets of models based on the evolutionary relationships inferred in the phylogenetic analysis with IQ-TREE and SVDQuartets (See results) were independently analyzed for (i) the Red Sea, (ii) the PAG plus the Sea of Oman, and (iii) the entire Arabian Peninsula. Three populations were used as lineage representatives for each set of models. The Brisbane population was included as outgroup in all models to calibrate times of divergence (He et al., 2019; He et al., 2020; X. Li et al., 2016). Although more comprehensive models including a higher number of populations could have been tested, we chose this approach to avoid the multiplicity of overly complex models resulting from the combination of the alternative scenarios for each one of the biogeographic regions. To avoid confounding effects from divergent evolutionary histories, we opted for a limited number of representative populations per geographic region instead of merging populations based on clustering and phylogenetic analyses (Hansen et al., 2018; Pedersen et al., 2018). For the Red Sea, the populations of Duba, Al Kharrar and Farasan Banks 2 (hereafter FB2) were included in three models with different topologies: simultaneous cladogenesis of the three lineages for a scenario of fast diversification; dichotomic cladogenesis from north to south; and a third scenario in which Al Kharrar from central Red Sea would have originated by admixture of the boreal and meridional populations. Because the Red Sea was never completely drained through the glacial cycles during the last 400,000 years, no assumptions were made about putative ancestral barriers to gene flow and coalescence times were allowed to vary freely. In the case of the PAG, two general hypotheses for lineage differentiation were tested: a scenario of recent differentiation following the colonization of the PAG after the last glacial maximum (LGM); and a scenario of early lineage diversification in which populations within the PAG became geographically isolated in glacial refugia. To test these two competing hypotheses, we fixed the coalescence times either to 700 generations or less (14,000 years); or to above 900 generations (18,000 years) assuming a generation time of 20 years (X. Li et al., 2016). Representative populations included in the model were Dammam from the northern basin, Ras Ghurab from the southern basin, and Shinas from the Sea of Oman. Three different tree topologies were modeled: a case of simultaneous cladogenesis corresponding to a fast diversification process; and two more models in which the most recent split corresponded either to Dammam-Ras Ghurab or to Ras Ghurab-Shinas. Each one of these topologies was tested under the postglacial colonization and divergence in glacial refugia scenarios. In the test for the entire Arabian Peninsula, the populations of FB2, Taqah and Ras Ghurab were used as representatives of the Red Sea, the Arabian Sea and the PAG, respectively. A single topology matching the IQ-TREE and SVDQuartets phylogenies was tested, and divergence times were allowed to vary freely. Time of coalescence of the Arabian lineages with Brisbane was set to 2.7 million years ago in all models (X. Li et al., 2016). Every model was compared under two different gene migration scenarios: a ‘strict isolation’ scenario with migration rates set to zero; and an ‘isolation with migration’ scenario where migration rates could vary freely (Table S4, Fig. S1, Supporting Information). As input data we used the folded site frequency spectra (SFS) generated from resequencing data. We retained the samples corresponding to the groups of study of each of the analyses from the ‘Full Dataset’. In these analyses, SNP loci under linkage disequilibrium were filtered out applying and less strict r2 limit of 0.4 in windows of 10K bp. A HWE filter for SNPs with a p-value of 10-4 was implemented. Because singletons are important for estimating parameters and likelihoods, no MAF filters were applied. Positions with less than 50% of individuals genotyped for each taxon/population were removed from the data matrices. Final matrices were of 58,613; 72,119 and 69,113 SNPs for the Red Sea, the PAG and the entire Arabian Peninsula analyses, respectively. The SFS were generated with easySFS version 0.0.1 (https://github.com/isaacovercast/easySFS; Gutenkunst, Hernandez, Williamson, & Bustamante, 2009) maximizing the number of segregating sites as recommended by the author (pers. comm.). Parameters with the highest likelihood were estimated under each of the models after 50 cycles of the algorithm, with 150,000 coalescent simulations per cycle. This procedure was replicated 100 times and the set of parameters with the highest final likelihood was retained as the best point estimate. To identify the model that better fit the data, we applied the Akaike information criterion (AIC; Akaike, 1998). For estimating 95% CIs of the parameters under the best model, we applied a non-parametric bootstrap procedure. Bootstrapping was carried out by splitting the SNP matrices in 100 SNP blocks and randomly combining them for each one of the repetitions. A total of 100 analyses were run for confidence interval estimation. TreeMix version 1.13 (Pickrell & Pritchard, 2012) was used to model historical patterns of gene flow between mangrove populations. The corresponding SNP dataset was built applying the same filters as for heterozygosity and diversity analyses, with the exemption of linkage disequilibrium filters (SNP matrix = 797,949) as it can be controlled for in the TreeMix command line. We ran TreeMix for 0–15 migrations, grouping SNPs in blocks of 50. Migration edges were plotted until 99.8% of the variance in ancestry between populations was explained by the model (Pickrell & Pritchard, 2012). The consistency of migration edges was evaluated by running TreeMix with 50 total replicates for each added migration edge number using a different, randomly generated seed. Results from the seed that yielded the highest likelihood are reported. Candidate gene identification with genotype-environment association analysisWe used genotype-environment association (GEA) analysis to identify candidate genes evolving under specific environmental pressures, and to estimate their contribution to patterns of local adaptation in mangroves from the Arabian Peninsula. We applied a redundancy analysis approach (Borcard, Gillet, & Legendre, 2011; Legendre & Legendre, 1998; Van Den Wollenberg, 1977) as implemented in the vegan R package. As explanatory variables, we used georeferenced environmental data extracted for the coordinates corresponding to the sampling sites and averaged over populations. Despite the availability of remote sensing data for a high number of both terrestrial and marine environmental parameters, we opted for a hypothesis oriented, ecologically informed approach to build our dataset of explanatory variables. Environmental parameters presenting particularly variable and extreme gradients across the Arabian Peninsula were specifically selected according to their relevance to mangrove ecology (Norman Duke, Ball, & Ellison, 1998; Naidoo, 2016). This dataset included two marine variables from the MARSPEC database (Sbrocco & Barber, 2013) approximating maximum of sea surface salinity and minimum of sea surface temperature averaged over months (MS_biogeo10_sss_max_5m, MS_biogeo14_sst_min_5m), two parameters of particular importance for mangrove physiology (Hogarth, 2015). It also included four terrestrial parameters from WorldClim2 (Fick & Hijmans, 2017; Hijmans, Cameron, Parra, Jones, & Jarvis, 2005) consisting of isothermality (WC_bio3), quantifying how the range of day-to-night temperature differs from the range of summer-to-winter, a variable generally useful for tropical and maritime environments (Nix, 1986); maximum air temperature of warmest month (WC_bio5) and minimum air temperature of coldest month (WC_bio6), both relevant ecological extremes in the Arabian habitats (Martínez‐Díaz & Reef, 2022); and annual precipitation (WC_bio12) as a proxy of aridity (Table S5, Supporting Information). Minimum sea surface and air temperatures showed high correlation, and the latter was removed when applying a correlation cutoff of 0.75. Annual precipitation was also filtered out by applying a forward selection method, implemented with an adding p-value limit < 0.05 and 1,000 permutations (Blanchet, Legendre, & Borcard, 2008; Capblancq, Luu, Blum, & Bazin, 2018). The remaining variables were retained. As response variables, we used population allele frequencies at each variant position. We chose to analyze population frequencies over individual genotypes because environmental scores were nearly identical for individuals from each population, given the spatial resolution of the remote sensing data. After excluding Brisbane samples from the ‘Full Dataset’, the HWE and MAF filters previously used were applied. Positions with less than 75% of individuals genotyped for each population were removed for a final dataset of 2,488,560 SNPs. Allele frequencies were then computed over populations to be used as the matrix of response variables. The genotyping quality filters employed here are commonly utilized in GEA studies based on RDA (e.g. Brauer, Hammer, & Beheregaray, 2016; Chang, Fridman, Mascher, Himmelbach, & Schmid, 2022; Faske et al., 2021; Friis et al., 2022; Friis et al., 2018; Laporte et al., 2016; Ortiz et al., 2023; Ruiz Miñano et al., 2022; Vu et al., 2020). However, overly conservative quality filters may limit outlier detection analyses based on genotype-environment association analysis. To investigate the impact of these filters in our GEA test, we conducted the analysis here described using an SNP dataset with less stringent quality filtering parameters.Two GEA analyses were implemented: a simple redundancy analysis (RDA) to test for genotype-environment associations between allele frequencies and environmental predictors; and a partial redundancy analysis (pRDA), in which in addition, we controlled for population structure effects. Covariates accounting for population structure consisted of the first two PCs of a PCA based on the allele frequencies matrix after filtering out positions putatively under selection, identified using the approach previously described with PCADAPT. Following the procedure described in Capblancq et al. (2018), we used the redundancy analyses to identify candidate genes potentially involved in divergent selection based on Mahalanobis distances estimated between each SNP and the center of the RDA space (Capblancq & Forester, 2021). Controlling for population structure is a common approach to reduce the rate of false positives due to historical processes. However, it may be overly conservative when neutral genetic variation correlates with environmental divergence, resulting in a higher frequency of false negatives and reduced power to detect signals of selection associated with environmental parameters (Ahrens et al., 2021; Capblancq, Lachmuth, Fitzpatrick, & Keller, 2022). Furthermore, in a series of comparative analyses using simulated data in a range of different demographic scenarios, Forester, Lasky, Wagner, and Urban (2018) found that controlling for distance/population structure when using RDA for GEA analysis led to a slight increase of the false positive rate. In our study, including covariates to account for population history effects resulted in a considerable increase in the number of detected candidate loci (see results), suggesting that this approach may be introducing distorting effects leading to an increase of the false positives rate. To control for associations signals resulting from covariation between neutral differentiation and ecological variation when identifying candidate loci, but also avoid potential false positives derived from correcting for population structure, only SNP outliers detected in both the simple and the partial RDAs were examined. A p-value threshold of < 0.01 was used for significance, and the Bonferroni correction for multiple tests was used to adjust the p-values. Based on the amount of explained variance in each one of the models, two and four RDA axes were used for identifying candidate loci in the simple and partial RDAs, respectively (See results). Then, we used the annotation of our reference genome (Friis et al., 2020) to survey our set of candidate loci and identify those located within functionally annotated genes. In addition, to explore differential association patterns of candidate loci at the individual level, we conducted a complementary, simple RDA with the individual genotypes of the SNP outliers as response variables. ResultsPopulation structure and genetic diversity analysesA principal components analysis (PCA) revealed marked levels of population structure. A plot of the first two axes recovered a pattern of clustering that matched the geographic distribution of the sampled populations. Populations from the Red Sea and the PAG clustered apart, showing high overlapping within each biogeographic region. Populations along the coasts of the Arabian Sea also showed marked genetic structure: populations at the west (Salalah and Taqah) grouped together and apart from the remaining genetic clusters, while populations from the Sea of Oman at the east (Shinas and Qurm) clustered close to the PAG. The intermediate population of Filim occupied a central position along the PC2. The third and fourth axes also revealed marked differentiation within biogeographic regions. Northern and southern basins of the PAG clustered apart along PC3, as well as populations from the Arabian Sea, yet to a lesser extent. Populations of the Red Sea differentiated along PC4 (Fig. 2A). A SNMF analysis recovered variability patterns consistent with the PCA results. At K = 2, populations clustered into two groups separating the Red Sea from the Arabian Sea and the PAG. The plot for K = 3 also revealed the more isolated, adjacent populations from the west of the Arabian Sea (Salalah and Taqah) as a differentiated group, with populations along the northeast Oman coast and into the PAG showing decreasing degrees of shared ancestry. At K = 4, populations from the northern basin of the PAG (Bahrain and Dammam) appeared as a differentiated cluster. At K = 5, populations from northern Red Sea (Duba, Al Wajh 1 and Al Wajh 2) also appeared as a genetically differentiated cluster, yet showing a signal of coancestry decreasing with latitude with proximal populations from the central Red Sea (Al Kharrar and King Abdullah Economic City, hereafter KAEC) and to a lesser extent, Al Lith. In the plot for K = 6, populations from the Sea of Oman, Qurm and Shinas, clustered together with Filim, yet Shinas showed intermediate levels of shared ancestry with the genetic cluster of the southern PAG basin, which included Ras Al Khaimahh (hereafter RAK), Umm Al Quawain (hereafter UAQ), Ras Ghurab and Suweihat. At K = 7, populations from southern Red Sea (Al Lith; Farasan Banks 1, hereafter FB1, and FB2) clustered apart from the central Red Sea populations, yet Al Lith presented intermediate levels of coancestry with the central Red Sea population KAEC (Fig. 2B). A Mantel test based on putatively neutral, independent SNP loci revealed a significant correlation between by-sea pairwise distances and Nei’s genetic distances among sampled populations (r = 0.765, p-value < 10-4; Table S6, Supporting Information). A linear regression between geographic distances and Nei's genetic distances showed a highly significant correlation pattern consistent with the results of the Mantel test (r2 = 0.58, P = 3.82x10-34, Fig. S2, Supporting Information). Pairwise FST values were all significant (p-value < 0.001) and ranged from 0.0140 to 0.1609, with the highest value observed between Northern Red Sea and Arabian Sea populations. Populations within the Red Sea, and within the PAG plus the Gulf of Oman showed lower levels of differentiation compared to those between biogeographic regions (Table S7). A two-level AMOVA was conducted to investigate the genetic structure of Arabian mangroves. The results showed differences between phylogenetic clades accounting for 9.45% of the total explained variance. Differences between populations accounted for 15.72 % of the explained variance, with the largest part of variance (74.83%) remaining within populations. ΦCT (0.095) and ΦSC (0.174) values revealed that structured genetic variation occurs both among clades and among populations within clades, yet ΦST (0.252) revealed that most of the genetic variation is found among populations overall (Table 2). Heterozygosity and nucleotide diversity indices revealed overall similar levels of genetic variability in Arabian mangroves. Populations with higher levels of shared ancestry at Al Kharrar, KAEC, Filim, Qurm and Shinas as inferred in the SNMF analyses, generally showed higher diversity scores (Table 1).Phylogenetic analysisA maximum likelihood phylogenetic reconstruction was consistent with the PCA and SNMF analyses, showing marked differentiation between and within biogeographic regions, with almost all the populations sampled resolved in clades with high node support. Arabian mangrove populations grouped in three major, reciprocally monophyletic clades or phylogenetic lineages: the Red Sea lineage (Major Clade I), the southern Arabian Sea lineage (Major Clade II) and the PAG plus the Sea of Oman lineage (Major Clade III) (Fig. 3). Within the Red Sea’s Major Clade I, the phylogenetic analysis revealed a sequence of cladogenetic events consistent with the geographic distribution of the sampled populations, with the first split separating northern (Duba, Al Wajh 1 and Al Wajh 2) and central populations (Al Kharrar and KAEC) from those of the south (Al Lith, FB1 and FB2); adjacent Al Kharrar and KAEC populations from central Red Sea showed little differentiation and grouped into a single clade. The Arabian Sea’s Major Clade II encompassed the populations Salalah, Taqah and Filim, all occurring in southern Oman, and appeared as the sister group of the Major Clade III, which includes all Sea of Oman and PAG populations. Major Clade III was further divided into two clades separating the populations of the northern basin of the PAG (Bahrain and Dammam) from those of the southern basin (RAK, UAQ, Ras Ghurab and Suweihat) and the Sea of Oman (Qurm and Shinas). Populations of these two latter subregions, which occur on each side of the Strait of Hormuz, appeared as reciprocally monophyletic sister groups (Fig. 3).The phylogenetic tree obtained with SVDquartets recovered the same major clades as in the IQ-TREE analysis, yet showed some variations in population grouping, with a poorer fit with the geographic distribution of the analyzed populations. Specifically, in the Red Sea, northern populations (Duba, Al Wajh 1, and Al Wajh 2) formed a sister clade to the southern populations (Al Lith, FB1, and FB2), with the clade formed by Al Kharrar and KAEC populations from the central Red Sea as the external group. Within the PAG, UAQ appeared as the external group to the remaining populations of the southern basin (RAK, Ras Ghurab, and Suweihat) instead of RAK. All nodes in the phylogeny received 100 bootstrap support (Fig. S3, Supplementary Information). Population and demographic history of the Arabian gray mangroveDemographic models were compared under fastSIMCOAL2. In the Red Sea, the model with the greatest likelihood score (ΔAIC = 318, Fig. 4; Table S8, Supporting Information) revealed a single cladogenetic event for the three analyzed populations under an isolation with migration scenario, and a splitting time dating back to 99,200 years (95% CI = [40,062 – 295,152]), consistent with a process of rapid, simultaneous lineage differentiation. Gene exchange among lineages was particularly high from Al Kharrar in the central Red Sea towards FB2 at the southern entrance (MIG Al Kharrar → FB2 = 60, 95% CI = [13 – 109] migrants per generation). For the remaining migration bands, 95% confidence intervals varied by orders of magnitude and included values near to zero, revealing little impact of these parameters in the SFS (Fig. 4; Table S8, Supporting Information). In the case of the PAG plus the Sea of Oman, the model that best fitted our data (ΔAIC = 1,615, Fig. 4; Table S8, Supporting Information) also revealed a scenario of simultaneous cladogenesis in isolation with migration dating back 37,760 years (95% CI = [18,950-161,565]), prior to the last glacial maximum. Signs of some gene flow were detected among PAG lineages. While narrower than in the Red Sea estimates, the ranges also included values near to zero in most cases, reducing the certainty in the model (Fig. 4; Table S7, Supporting Information). In the analysis with representatives of each biogeographic region of the Arabian Peninsula, the highest likelihood corresponded once again to the model including migration (ΔAIC = 1,067, Fig. 4; Table S8, Supporting Information), and revealed splitting times of 70,180 years (95% CI = [14,074-161,976]) and 153,140 years (95% CI = [90,714-416,863]) (Fig. 4; Table S8, Supporting Information). We used TreeMix (Pickrell & Pritchard, 2012) to further explore patterns of historical gene flow among Arabian mangrove populations. Phylogenetic relationships recovered with TreeMix were consistent with the maximum likelihood tree generated with IQ-TREE. A tree model with ten migration events explained up to 99.8% of the SNP variance, revealing signs of both historical and contemporary gene flow among mangrove populations. Two recovered bands of ancestral migration between the outgroup lineage of Brisbane and the northern populations of both the Red Sea and the PAG seemed highly unlikely, so that these results should be interpreted with caution (Fig. 5). Candidate genes and environmental adaptation patterns in Arabian gray mangrovesWe applied simple and partial redundancy analyses to identify candidate loci potentially involved in differential selection among Arabian gray mangrove populations. Environmental predictors explained 33.2% of the total genetic variance in the simple RDA (Adjusted R2 = 0.332, p-value < 0.01). The scores of the simple RDA revealed distinct association patterns among biogeographic regions. Red Sea mangroves showed high, negative loading values on RDA axis (RD) 1, while the remaining populations correlated positively with this axis, and heavily in the case of the Sea of Oman and PAG southern basin. In contrast, the Arabian Sea populations showed high negative association values with RD2, while Red Sea and PAG mangroves showed positive association scores along this axis, yet the former with much lower correlation values. In the pRDA, environmental variables explained 6.4%of the total genetic variance after controlling for population structure (Adjusted R2 = 0.064, p-value < 0.01). Scores revealed a more scattered pattern of variation among and within biogeographic regions, where populations from northern PAG showed high positive correlation with pRD1, while populations of the southern PAG and Arabian Sea, as well as from Central Red Sea, showed more limited, negative loading values. In the case of the pRD2, highest loading values corresponded to Qurm, with a positive correlation, and to Ras Ghurab and Suweihat, that correlated negatively (Table S9, Supporting Information). The simple and the partial RDA were jointly used to test for signs of selection applying the method described in Capblancq et al. (2018). The first to axes of the simple RDA accounted for an unadjusted proportion of explained variance equal to 38.8% (R2 = 0.388) out of 48.0% explained by the full model. In turn, the four axes of the partial RDA accounted for 32.3% (R2 = 0.323). Two and four RDA axes from the simple and partial RDA, respectively, were thereby used for candidate loci identification. The analyses revealed 446 genetic variants showing significant associations with one or more of the four environmental predictors in both the RDA and pRDA (NRDA = 3,015; NpRDA = 73,671; Njoint = 446; Fig. S4, Supporting Information). Of the 446 outliers, 70 were located within 31 annotated genes with known functions or associated gene ontology terms (Table S10, Supporting Information). To visually explore how adaptive variability is structured at the individual level, we conducted a complementary redundancy analysis based on the individual genotypes of the 446 candidate loci alone. The plot of the first two RDA axes revealed distinctive associations patterns only partially structured by biogeographic region: Populations from the Red Sea differentiated along a gradient loading on maximum air temperature, with northern populations (Duba, Al Wajh 1 and Al Wajh 2) showing high negative correlation values, central Red Sea (Al Lith, FB1 and FB2) clustering close to the origin of coordinates, and southern Red Sea showing positive correlations. Populations from the southwest of the Arabian Sea (Taqah and Salalah) showed a strong, positive GEA signal with isothermality, while correlated negatively with a maximum salinity gradient. In contrast, populations from the Sea of Oman presented low association values and clustered around the origin of coordinates. Populations from the PAG showed an opposite GEA pattern to those of the Arabian Sea, also differentiating along the salinity/isothermality gradient, yet associations were weaker and more scattered in the southern basin than in the northern one (Fig. 6A). Individual Euclidean distances averaged over populations in the RDA ordination space showed general similar trends, with within-region, highest differences among association patterns occurring along the Red Sea and outrunning divergence between biogeographic regions (Fig. 6B). The results of the RDA conducted to investigate the impact of the filters applied in our GEA test using more lenient thresholds are reported in the Supporting Information (Table S11). DiscussionModerate genetic structure and robust phylogenetic support suggest a process of lineage diversification consistent with IBD and geographic isolation Population structure analyses revealed moderate yet marked levels of divergence across Arabia. Both a PCA and a SNMF analysis based on putatively neutral SNPs recovered signs of widespread differentiation, and identified groups of genetic clusters that mirrored the geographic distribution of mangroves across the various seas surrounding the Arabian Peninsula. The analyses also showed genetic differentiation between the populations from the north, the center and the south of the basin of the Red Sea; as well as between the northern and southern basins of the PAG. Considerable levels of shared ancestry between the populations of the southern PAG basin and the Sea of Oman were also detected, suggesting either incomplete lineage sorting or ongoing gene flow between them.A Mantel test revealed a significant correlation between neutral genetic differentiation and geographic distance among populations. However, a linear regression showed values of genetic Nei’s distance that did not always increase monotonically with geographic distance. While a steep linear correlation was observable among populations within the Red Sea and the PAG, this relation was less pronounced across regions, suggesting that drift has been more influential than gene flow shaping patterns of genetic differentiation at greater distances and between sets of populations separated by geographic barriers (Hutchison & Templeton, 1999). Despite of this general pattern, two groups of comparisons showing negative and positive residuals corresponding to within and between basins, respectively, were observed in the PAG, suggesting some degree of isolation decoupled from geographic distance also within regions. These results support the role of IBD in the differentiation of mangrove populations, yet they also suggest that geographic barriers to gene flow have likely contributed to differentiation at different geographic scales. However, because barriers are sparsely distributed along the geographic distribution of the Arabian mangroves, distinguishing between IBD and divergence in geographically isolated populations is statistically challenging in this particular system.Phylogenetic inference revealed a general pattern of reciprocal monophyly among Arabian mangrove populations, and identified differentiated phylogenetic lineages occurring in the Red Sea, the Arabian Sea and the PAG, with the latter including the populations of the Sea of Oman. Patterns of phylogenetic divergence among Arabian biogeographic regions have been previously reported for other marine taxa (DiBattista et al., 2020; Ketchum et al., 2020; Saenz‐Agudelo et al., 2015; Smith et al., 2022; Smith, Hume, Delaney, Wiedenmann, & Burt, 2017), congruent with the geographic distribution of the species and with physical barriers to gene flow (DiBattista, Roberts, et al., 2016; DiBattista et al., 2020). However, few marine organisms in Arabia exhibit comparable levels of differentiation as the gray mangrove at a within-region geographic scale, (e.g. DiBattista et al., 2020; Ketchum et al., 2020; Saenz‐Agudelo et al., 2015; Smith et al., 2017; Torquato et al., 2022; Torquato et al., 2019), suggesting a process of population differentiation occurring within the historically enclosed water bodies of the Red Sea and the PAG. Neutral population structure and phylogenetic relationships here reported support the hypothesis of mangroves finding refuge within the enclosed water bodies of the Arabian Peninsula during Pleistocene glacial cycles, where lineages could diverge in long-term geographic isolation. Geological and paleoclimatic data suggest that areas of the Red Sea basin could indeed have acted as glacial refugia for mangroves (Lambeck et al., 2011), as it has been proposed for other endemic marine lineages of the region (DiBattista, Choat, et al., 2016; DiBattista, Roberts, et al., 2016). This scenario is particularly striking in the case of the PAG, where the presence of potentially suitable habitats was arguably very limited during glacial periods, where only the estuary of the historic Shatt-Ur river outlet to the Sea of Oman near the Strait of Hormuz had been thought to occur (Lambeck, 1996). Interestingly, populations from the southern basin of the PAG appeared as more closely related to the populations of the Sea of Oman, on the outside of the Strait of Hormuz, than with those of the northern PAG basin. This sequence of cladogenetic events also better fits a scenario of differentiation in glacial refugia where populations in the north would have become isolated earlier as the shore lines retreated, as opposed to a scenario of recent colonization throughout the PAG after the LGM, where northern populations would represent the most recent phylogenetic split (e.g. Smith et al., 2022). Demographic inference supports lineage divergence in (cryptic) glacial refugiaA total of 20 demographic models were compared with fastSIMCOAL2 to identify the most likely scenario under which mangroves from the Red Sea, the PAG and the Arabian Peninsula overall, diversified. For the Red Sea, AIC revealed that the most likely scenario was consistent with a simultaneous cladogenesis occurring during a period spanning the three last glacial cycles, suggesting that mangroves may have colonized the Red Sea basin shortly after the Strait of Bab al-Mandab opened to the Indian Ocean ca. 400,000 years ago (Lambeck et al., 2011) and persisted thereafter. Glacial cycles in the Red Sea are characterized by changes in the eustatic sea level and connectivity, but also by periods of desiccation and sharp changes in salinity and temperature. Therefore, the results reported here suggest that the levels of population and phylogenetic diversity found in the Red Sea are the product of historical events involving changes in aspects of population history such as effective size and geographic isolation, but also in environmental conditions (DiBattista, Choat, et al., 2016; DiBattista et al., 2020). In the case of the PAG, the model that better fit our data revealed a scenario of simultaneous cladogenesis dating to nearly 40K years ago, prior to the LGM and the later flooding of the enclosed sea, further supporting the presence of cryptic glacial refugia within the PAG suggested by our phylogenetic analyses. Drastic changes in environmental conditions during glacial maxima, particularly in terms of salinity and temperature may have presented exceptional physiological challenges to resident marine life. Until now, survival through glacial periods has not been documented for any marine taxa in the PAG. However, conditions of intertidal zones in marine swamps, while extreme, may have remained locally suitable for gray mangroves, which occur in a wide range of salinities globally. Lambeck (1996) reconstructions of historic shorelines at times of the LGM show the potential development of lagoons and lakes in several localities, with the main ones corresponding to what are now northern and southern basins, as well as in the Strait of Hormuz. Indeed, today A. marina occurs in a number of inland, non-tidal saline (e.g. Ellison & Simmonds, 2003; Stoddart, Bryan, & Gibbs, 1973) or even freshwater lakes (Beard, 1967; Taylor, 1986) under conditions that could resemble those of the PAG during glacial periods. Also, previous examples of marine cryptic glacial refugia have been documented for taxa such as seaweeds, rays and snails in places like southwest Ireland, southwest Greenland, the northern Brittany-Hurd Deep area of the English Channel, the Azores islands or the northwest Iberian Peninsula (Bringloe et al.; Chevolot, Hoarau, Rijnsdorp, Stam, & Olsen, 2006; DiBattista, Choat, et al., 2016; Hoarau, Coyer, Veldsink, Stam, & Olsen, 2007; Provan, Wattier, & Maggs, 2005), suggesting that the persistence of benign local conditions in marine environments during glacial periods may be relatively common. In addition to a split occurring prior to the LGM, the best fitting demographic model revealed potential gene flow between PAG basins and between the southern basin and the Sea of Oman. The TreeMix analysis also detected significant levels of gene flow between both terminal and ancestral branches, suggesting that some migration could occur as mangrove populations became isolated and also after the LGM, once the infilling of the PAG started. Potential barriers such as the narrow Strait of Hormuz or the marine eddies between Qatar and Iran separating the northern and southern basin may, therefore, not be as limiting for mangrove propagule dispersal as has been proposed for other marine-associated, non-plant species (Hoolihan, Premanandh, D’Aloia-Palmieri, & Benzie, 2004; Smith et al., 2022). This contrasts, however, with higher FST and Nei’s genetic distance scores among populations of the northern and southern PAG basins, suggesting that dispersal events may have been limited, and that these results may need to be further tested.The highest likelihood model for the entire peninsula supported a recent common evolutionary origin and for the mangroves of Arabia. The model was congruent with a process of diversification occurring over the different biogeographic regions of the peninsula, arguably driven by geographic isolation and large-scale changes in environmental conditions and connectivity resulting from the glacio-eustatic cycles of the Pleistocene. Coalescence time between the representative populations of the PAG (Ras Ghurab) and the Arabian Sea (Taqah) dated back 70K, lending support the hypothesis of a lineage split prior to the LGM. The splitting time between the Red Sea (represented by FB2) and the PAG/Arabian Sea ancestral lineage dates back to, at least, the onset of the last glacial period and potentially and at most, to times close the opening of the Red Sea to the Indian Ocean. Both fastSIMCOAL2 and TreeMix analyses detected a certain degree of gene exchange among ancestral and terminal branches, suggesting a lack of reproductive barriers among lineages upon secondary contact.Our analyses reveal a process of lineage diversification in the gray mangroves of the Arabian Peninsula that spanned the last two to three glacial periods. Generation time for the gray mangrove has been estimated at 20 years in tropical areas (He et al., 2019; Triest et al., 2021), a value that we retained in our analyses as calibrating of our demographic models was based on a splitting time estimate using this value (Li et al., 2016). However, generation time in high-latitude populations of the PAG have been reported to range from 25.6 to 53.8 years (Ali, Alfarhan, Robinson, & Aldjain, 2008), which implies that the diversification process may date back as much as twice as early as inferred in our analyses. Modelling divergence times in recently diversified systems has proven challenging, even with large datasets of genome-wide markers (Beichman, Huerta-Sanchez, & Lohmueller, 2018). Lenient assumptions such as constant effective population size along branches, together with the wide confidence intervals estimated in fastSIMCOAL2, entail substantial uncertainty, requiring caution when interpreting the reported results. Additionally, it is important to note that population splits were not entirely consistent between demographic models and phylogenetic analyses, further emphasizing the need for careful interpretation. However, the observed patterns and extent of phylogenetic and neutral genetic structure in Arabian mangroves, together with the conservative generation time implemented in our models, largely support a diversification process occurring before the LGM. These findings strongly suggest divergence within glacial refugia as a key driver in the Arabian mangrove system. Nevertheless, these results should be revisited in the future, potentially employing analytical methods such as Approximate Bayesian Computation (ABC) and machine learning for demographic modelling.Although our results suggest that diversification took place before the LGM, the Arabian mangroves system represents a strikingly fast process of diversification, occurring at temporal scales of tens to a few hundred thousand years. Marked levels of population divergence are expected at the edge of species’ ranges, where reduced population sizes and pronounced habitat fragmentation can lead to rapid differentiation due to drift in isolation and divergent selection (Lesica & Allendorf, 1995; Schaal & Leverich, 1996). Indeed, higher genetic structure in the margins of the species distribution compared to core populations of the gray mangrove of the West Pacific has been previously reported (Arnaud‐Haond et al., 2006). Moreover, increased rates of speciation have been documented in other mangrove systems undergoing periodic cycles of isolation and gene flow due to fluctuations in sea level during glacial periods (He et al., 2019), similar to what may have occurred in the enclosed water bodies of the PAG and especially the Red Sea.Redundancy analyses reveal multi-loci adaptive divergence driven by environmental extremes In a simple and a partial RDA controlling for population structure, four constraining variables approximating critical environmental factors in mangrove ecology related to temperature extremes and hypersalinity, explained 33.2% and 6.4% of the total variance in population SNP frequencies, respectively. The amount of variance explained by environmental variables was comparable to the values reported in studies applying RDA to genotype-environment analysis in plants, typically ranging from 15% to 40% (e.g. Capblancq et al., 2020; Chang et al., 2022; Faske et al., 2021; Garot, Joët, Combes, & Lashermes, 2019; Leamy et al., 2016). A total of 446 candidate SNP outliers were identified based on their contribution to the genotype-environment association patterns. A complementary RDA based solely on the this set of SNP outliers and computed over individual genotypes revealed considerable adaptive divergence among Red Sea populations, which differentiated along a gradient of maximum temperatures from south to north. Differences in gene-environment correlation signals for populations of the PAG were less clear, yet the analysis recovered highly divergent association scores with respect to the populations of the west of the Arabian Sea along salinity maxima and isothermality gradients. These association patterns suggest that local adaptation has likely played a significant role in the differentiation of the mangrove lineages in the Arabian Peninsula (See Supporting Information for further details). We identified 31 functionally annotated genes associated with one or more RDA outliers. Among the functions associated to detected candidate genes, at least eleven seemed particularly relevant for adaptation to the extreme and variable habitats of the seas around the Arabian Peninsula. Mitochondrial GrpE (Mge) proteins act as molecular chaperones that assist in the refolding of proteins within the mitochondria, playing a key role in maintaining homeostasis, especially during stress conditions (Hu, Lin, Chi, & Charng, 2012; Mayer & Bukau, 2005). In Arabidopsis thaliana, the MGE2 gene has been shown to be induced by heat, and to be involved in tolerance to prolonged high temperatures (Hu et al., 2012). The abscisic acid-insensitive like2 (ABIL2) gene has been identified as a negative regulator of ABA signaling, a key stress phytohormone, and to positively regulate stomatal aperture and density in rice (C. Li, Shen, Wang, & Wang, 2015). Overexpression of ABIL2 results in modified plant developmental traits, such as altered stomatal density and root architecture, which may render plants more susceptible to drought stress (C. Li et al., 2015). Plasma membrane intrinsic (Pip) proteins, are a subgroup of aquaporins, which are membrane channel proteins involved in the transport of water and other small molecules across cell membranes (Agre, 2006). Pip proteins are crucial for regulating water movement during various physiological processes and regulating stomatal conductance (Sakurai, Ishikawa, Yamaguchi, Uemura, & Maeshima, 2005). PIP gene expression has been reported to mediate the response to drought and salt stress in various species, including barley, citrus, and tobacco (Katsuhara & Hanba, 2008; Mahdieh, Mostajeran, Horie, & Katsuhara, 2008; Rodríguez-Gamir et al., 2011). Beta-xylosidase genes such as BXL2 code for enzymes involved in the degradation of hemicellulose, a major component of plant cell walls, playing essential roles in remodeling its composition and architecture (Collins, Gerday, & Feller, 2005; Goujon et al., 2003). The cell wall is a crucial structure that serves as a barrier against environmental stresses. During periods of stress, such as drought, high salinity or pathogen attacks, plants undergo adaptive responses in cell wall composition (Goujon et al., 2003; Zhao et al., 2010). The MOB1A gene codes for a kinase regulator that plays a pivotal role in tissue patterning and organ growth, and in A. thaliana serves a plant-specific function by contributing to growth modifications in response to stressful conditions (Pinosa et al., 2013). WRKYs represent one of the largest transcription factor families with relevant biological functions in response to different kinds of abiotic and biotic stressors in vascular plants (Jiang et al., 2017; Phukan, Jeena, & Shukla, 2016), including A. marina (Feng et al., 2023). In particular, the WRK50 here identified has been reported to play a role in regulating nitrogen use efficiency and coordinating plant responses to nitrogen levels and biotic stressors (Cheng et al., 2021). A study of the effect of salinity stress on gene expression in licorice revealed that the squalene synthase 2 (SQS2) was upregulated in leaves under high salinity conditions, suggesting a role for this gene in the response to salt stress (Shirazi, Aalami, Tohidfar, & Sohani, 2019). Squalene epoxidase (Sqe) enzymes are essential for the biosynthesis of sterols in plants. SQE1 and its homologues SQE2 and SQE3 transcripts can epoxidize squalene into oxidosqualene, the precursor of all known angiosperm cyclic triterpenoids (Rasbery et al., 2007). It has been proposed that SQE genes can play a role in the regulation of reactive oxygen species, stomatal responses and drought tolerance, likely mediated by the involvement of sterols in the localization of NADPH oxidases (Posé et al., 2009). The 4-coumarate-CoA ligase-like 7 (4CLL7) gene encodes an enzyme related to terpenoid metabolism potentially involved in response to drought stress, as it has been reported for red sage (Madritsch et al., 2019; Zhang, Li, Su, Song, & Wang, 2022). The abscisic acid-insensitive 5-like (AI5L5) gene is similar in function to ABI5, a well-known transcription factor involved in the response to the plant hormone ABA pathway. Under red-blue combined light, AI5L5 appeared as downregulated, and was likely involved bud flower differentiation in grape (Liu, Yuan, Dang, & Zhang, 2022). Finally, the PUMILIO (PUM) family is a group of genes that play important roles in various biological processes, including post-transcriptional regulation of gene expression. In A. thaliana, the PUM23 gene has been reported to mediate in the response to abiotic stresses, including salt stress in association with the ABA signaling pathway (Huang, Lin, & Cheng, 2018). Conclusion In this study, we used the Arabian mangroves complex to analyze the role of spatial environmental variation, geography and neutral evolution in driving population and lineage divergence. Phylogenetic analysis and demographic modeling revealed evolutionary relationships and times of population divergence consistent with periods of geographic isolation in glacial refugia both in the Red Sea and the PAG, ruling out the hypothesis of a postglacial colonization. A significant signal of IBD was detected as well, suggesting that geographic distance also played an important role in the differentiation of Arabian mangrove populations. Signs of multilocus local adaptation were detected among and within the phylogenetic lineages identified for each of the biogeographic regions, revealing significant environmental pressures driving adaptive divergence at different geographic scales. Broader geographic sampling across the entire gray mangrove distribution, nevertheless, will be needed to further investigate candidate functional loci involved in mangrove ecological divergence. Overall, the results reported in this study support a process of rapid diversification driven by the combined factors of environmental selection and historical events, and reveal peripheral mangrove populations of Arabia as potentially important sites for lineage diversification and sources of evolutionary innovation in response to environmental extremes.Acknowledgements The authors are grateful to NYUAD’s Core Bioinformatics for analysis and computational assistance. We thank Mark Priest, Dain McParland, Noura Al-Mansoori, Anique Ahmad, and Ada Kovaliukaite for their participation in the fieldwork. We gratefully acknowledge the Environment Agency Abu Dhabi (permit reference number 20181823a), and the Oman Ministry of Environment and Climate Affairs, Director General of Nature Conservation (permit number: 6210/10/75), for providing permits for research related to this manuscript. This work was supported by New York University Abu Dhabi Institute Grant (73 71210 CGSB9), NYUAD Faculty Research Funds (AD060) and King Abdullah University of Science and Technology baseline funding to CMD.