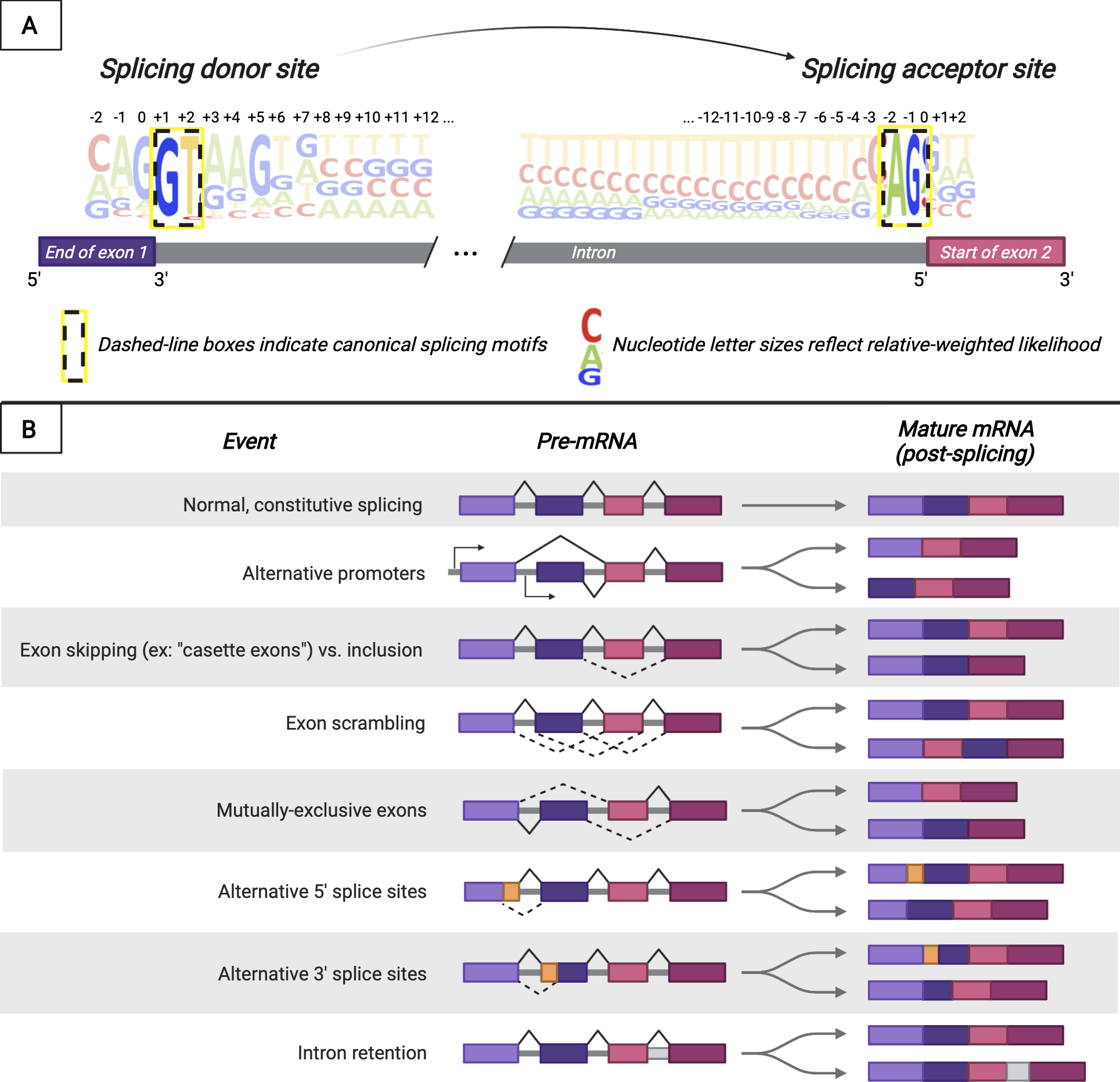
The vast volume of data that has been generated as a result of the next-generation sequencing revolution is overwhelming to sift through and interpret. Parsing functional vs. non-functional and benign vs. pathogenic variants continues to be a challenge. Out of three billion bases, the genomes of two given individuals will only differ by about 3 million variants (0.1%). Furthermore, only a small fraction of these are biologically-relevant and, of those that are functional, only a handful actually drive disease pathology. While whole genome and exome sequencing have transformed our collective understanding of the role that genetics plays in disease pathogenesis, there are certain conditions and populations for whom DNA-level data has failed to produce a molecular diagnosis. Patients of non-White race/non-European ancestry are disproportionately affected by “variants of unknown/uncertain significance” (VUS). This limits the scope of precision medicine for minority patients and perpetuates health disparities. VUS often include deep intronic and splicing variants which are difficult to interpret in DNA alone. RNA analysis is capable of illuminating the consequences of VUS thereby allowing for their reclassification as pathogenic vs. benign. Here we review the critical role, going forward, of transcriptome analysis for clarifying VUS in both neoplastic and non-neoplastic diseases.