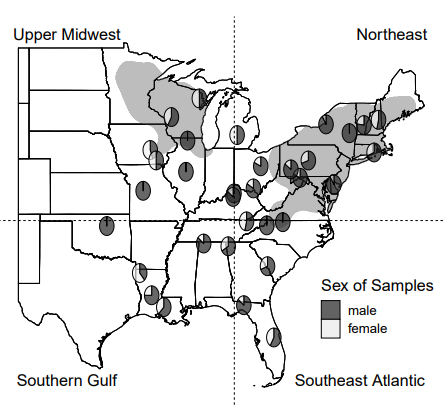
Tick vectors and tick-borne disease are increasingly impacting human populations globally. An important challenge is to understand tick movement patterns, as this information can be used to improve management and predictive modeling of tick population dynamics. Evolutionary analysis of genetic divergence, gene flow, and local adaptation provides insight on movement patterns at large spatiotemporal scales. We develop low coverage, whole genome resequencing data for 92 samples representing range-wide variation in blacklegged ticks, Ixodes scapularis, across the U.S. Through analysis of population genomic data, we find that tick populations are structured geographically, with gradual isolation by distance separating three population clusters in the northern U.S., southeastern U.S., and a unique cluster represented by a sample from Tennessee. Populations in the northern U.S. underwent population contractions during the last glacial period and diverged from southern populations at least 50 thousand years ago. Genome scans of selection provide strong evidence of local adaptation at genes responding to host defenses, blood-feeding, and environmental variation. In addition, we explore the potential of low coverage genome sequencing of whole-tick samples for documenting the diversity of microbial pathogens. Metagenomic analyses recover important tick-borne pathogens and their geographical variation, including the higher prevalence of Borrelia burgdorferi in northern populations, geographic strain variation in Rickettsia species, and the rare occurrence of B. miyamotoi and Anaplasma phagocytophilum. The combination of restricted pathogen distribution, isolation by distance, and local adaptation in blacklegged ticks demonstrates that gene flow, including recent expansion, is limited to geographical scales of a few hundred kilometers.