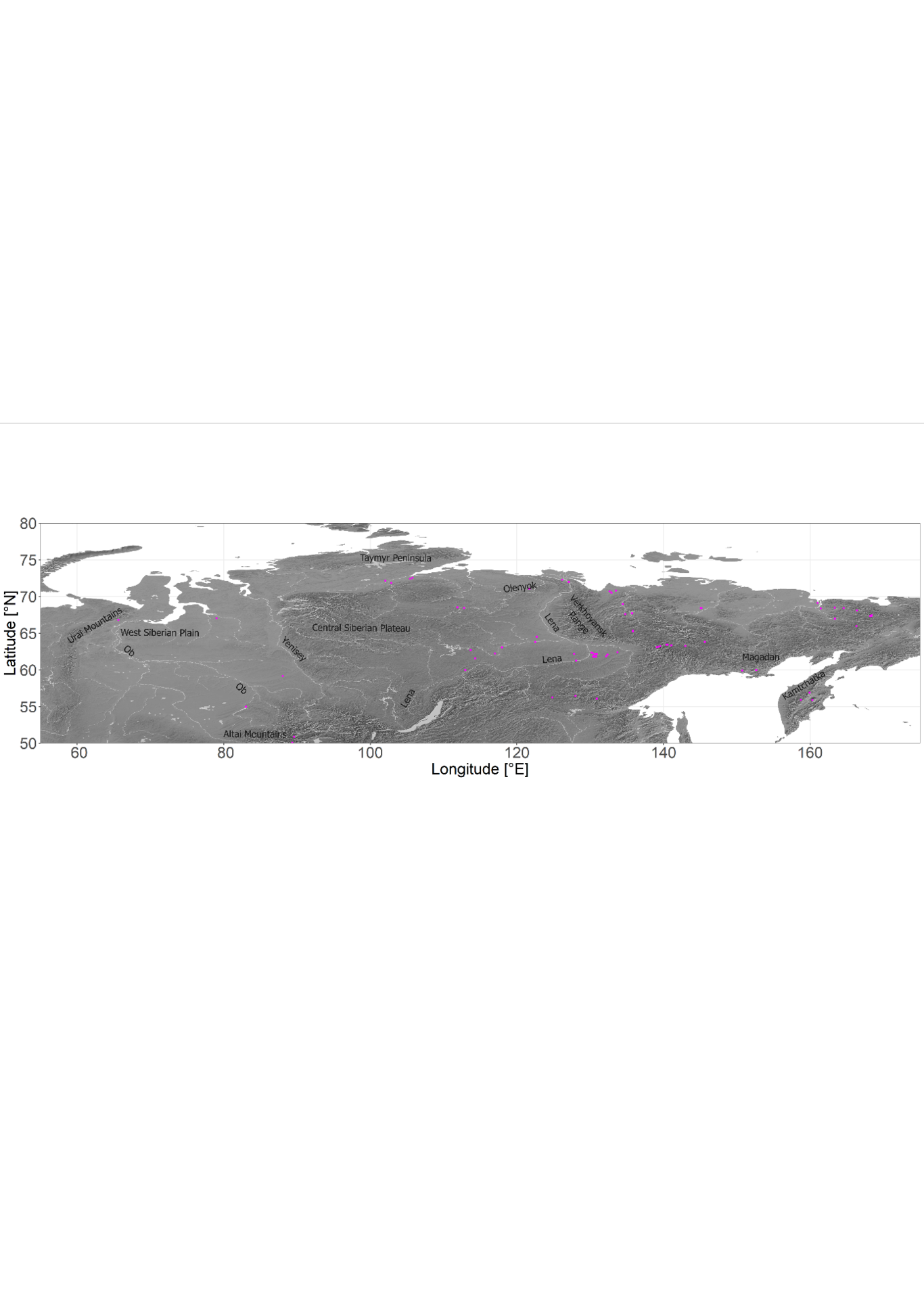
The present distribution of Siberian boreal forests that are dominated by larches is influenced, to an unknown extent, by the glacial history. Knowing the past treeline response to climate shifts can improve our understanding of future treeline dynamics under changing climate. Here, we study patterns in the genetic variability of Siberian larches (Larix spp.) that can help us to unravel biogeographic migration routes since the Last Glacial Maximum (LGM). We sampled 148 larch individuals from Eastern Siberia. For each individual, genome-wide single nucleotide polymorphisms (SNPs) were derived through genotyping by sequencing (GBS). We inferred the spatial distribution from 14,003 SNPs with a cluster analysis. To infer the postglacial demographic history of Larix, we applied an Approximate Bayesian Computation. The Bayesian population assignment statistically supported three to four clusters from Western to Eastern Siberia that correspond well to the geographic ranges of the main Siberian larch species Larix sibirica, L. gmelinii, and L. cajanderi. Using four plausible clusters, the tested hypotheses in DIYABC show that the existing populations seem to have been initiated long before the LGM. We presume that the different populations originate from larch populations that survived the glacial periods. From our genetic studies, we deduce that Larix was more likely to have survived the cold LGM in northern refugia, from where a fast colonization of Siberia was possible, rather than Larix completely repopulating Siberia in the postglacial spreading out from southern areas with less harsh climatic conditions. The northernmost expansion during the Holocene seems to have benefitted from refugial populations ahead of the treeline at that time, which explains the existence of Larix in the far north. We expect from our results that the present migration will be slow at first as there are currently no refugial populations far north, as there probably were in the Holocene.